嵌合抗原受体(CAR)T细胞已是肿瘤免疫治疗领域典型的范例,已成为肿瘤治疗历史上里程碑式的成就。2017年,FDA批准了两款针对CD-19 的CAR-T细胞治疗,两款CAR-T在B淋巴细胞瘤上有着显著地疗效。目前,多种CAR-T设计正在进行恶性血液病,乃至实体瘤免疫治疗的研究。为了在体外产生CAR-T细胞,慢病毒载体(LVs)由于其可以稳定的将大片段DNA有效的整合进分裂和非分裂细胞而被广泛应用。本文将综述生产CAR-T细胞的最新进展和挑战,以及用于制备GMP级别CAR-T 的LVs的生产过程,概述CAR-T治疗的应用进展,特别强调下一代同种异体CAR-T细胞。
近些年来癌症免疫治疗已经革命性的改变了肿瘤治疗手段,逐渐成为一种有效的低侵袭性的治疗策略,广泛的应用于多种肿瘤的治疗。多种策略正在研究用于癌症免疫治疗,包括过继性细胞治疗,细胞因子,单克隆抗体(mAbS), 以及肿瘤疫苗(图1)。
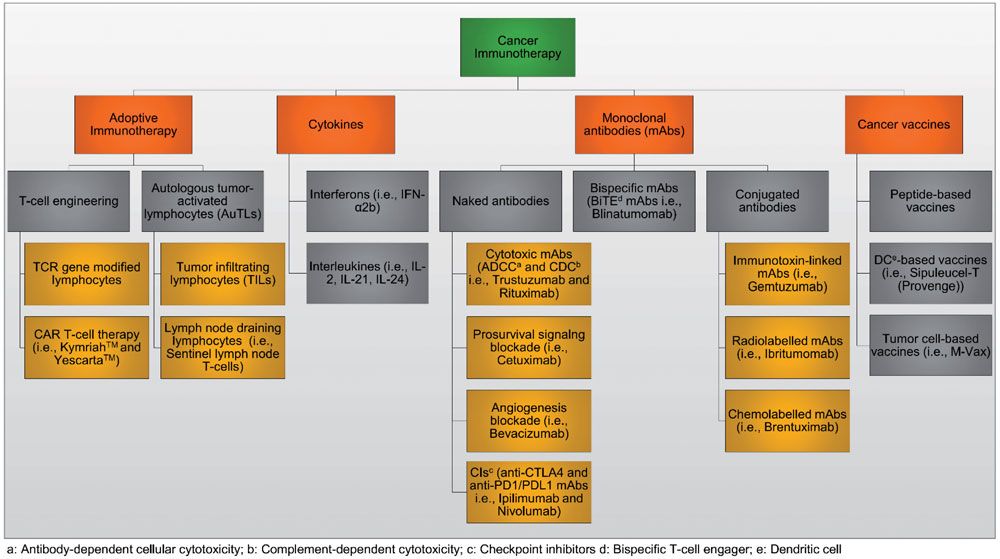
图1.肿瘤免疫治疗方法
在这些靶向肿瘤免疫治疗中。免疫检查点抑制剂和嵌合抗原受体(CAR)T细胞治疗具有令人振奋的响应率,在肿瘤免疫治疗领域取得了空前的成功,尤其是对那些其他方法治疗失败的患者来说,免疫治疗重新为他们的燃起了治疗希望。CARs是基因工程化的T细胞受体(TCRs),通常由胞外结构域和胞内结构域组成。其中胞外结构域包含一条源自单克隆抗体的单链可变区结构域(scFv),可以结合肿瘤细胞表面抗原。胞内结构域包含4-1BB(CD137)和/或CD28共刺激因子(为T细胞扩增和持久性提供第二信号),胞内结构域还包含TCR CD3ε链(为T细胞激活提供第一信号)。这些修饰的的TCRs设计使T细胞重定向,靶向癌症细胞特异性表面抗原。不同于自然地TCRs,CARs不依赖于主要组织相容性复合物(MHC),其主要作用类似于抗体检测细胞表面蛋白,糖磷脂,碳水化合物的表达。换句话来说CAR-T细胞的主要思想是将抗体特异性与抗原的结合的特性和T细胞胞内精确地细胞信号传导以及细胞杀伤作用有机的结合了起来(图2)。
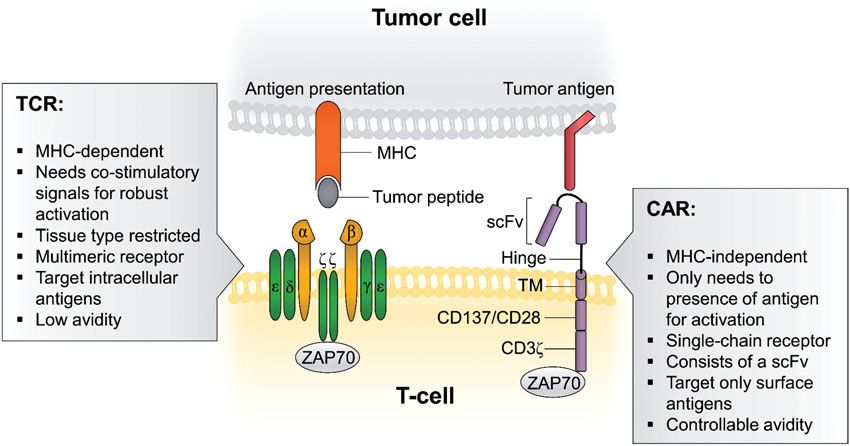
图2.TCR与CARs的不同
在CAR-T细胞治疗中,从病人自身分离到的T细胞,(无论是CD4+T细胞,还是CD8+T细胞,)通过基因工程化修饰,选择性的攻击肿瘤细胞。然而,CD8+ CAR-T(而不是CD4+ CAR-T)用于治疗容易导致T细胞耗竭,凋亡,有效性较差。另一种策略是通过鉴定分离肿瘤患者肿瘤组织中浸润性T淋巴细胞和外周血中的淋巴细胞,通过在体外选择,扩增,分化成为有效应功能,可增殖的肿瘤杀伤细胞。完全的阐述工程化TCRs在肿瘤免疫治疗中的应用超出了本文的内容范畴,但是其临床治疗效果有待商榷。两款已应用于临床治疗的CAR-T相比(KymriahTM(Novartis, Basel, Switzerland)and YescartaTM(Kite Pharma/Gilead Sciences, Foster City ),两款CD19 CARs都有着显著地抗血液肿瘤疗效,尤其是在难治的复发性B细胞急性淋巴性白血病治疗,以及难治性,复发大B细胞淋巴瘤治疗中,病人病情得到持续性缓解。KymriahTM 主要用于小儿和年轻患者复发性难治性前B细胞急性淋巴性白血病,YescartaTM 主要用于成年患者的B细胞霍奇金淋巴瘤。
病毒和非病毒载体都可用于CAR-T细胞生产。在病毒载体中,慢病毒载体(LVs)由于其有效的将基因整合进靶细胞基因组,稳定的基因表达等特点,被认为是最有希望的基因治疗载体。图3阐述了基于慢病毒载体LV的CAR-T细胞制备的主要步骤。与其他病毒载体相比,慢病毒载体的整合模式具有更低的致癌风险,以及随机整合基因风险低,因此,用慢病毒载体生产CAR-T细胞更加安全有效,而且灵活。更重要的是慢病毒相较于其他病毒载体,生产成本相对较低。另外,慢病毒载体可以转导分化细胞,也可以转导分化细胞,因此,慢病毒载体可以转导更为广泛的细胞,包括那些难转导的血液前体细胞,神经细胞,淋巴细胞和巨噬细胞。
本文主要描述目前已有的CAR-T类型和应用,以及以LV为载体的GMP级别的CAR-T生产。根据已发表文献,专利,临床治疗CAR-T,讨论基于LV的CAR-T生产策略。
CAR-T细胞产生
近年来,多种CAR免疫治疗策略正在改善着CAR-T治疗的有效性和安全性。在第一代CAR分子中,胞外域包含scFv结合于肿瘤相关抗原TAA,胞内结构域包含CD3ε信号结构域或Fc γ受体。在该结构中,CAR-T 激活第一信号由scFv结合TAA提供,CD3ε提供了T细胞激活的胞内信号,第2信号。第二代CAR-T中,实现了T细胞一步激活,CAR分子的胞内部分和CD3ε,共刺激分子CD28或4-1BB胞内信号结构域结合,信号1和信号2都由scFv与TAA的结合诱导刺激产生。尽管在小鼠抗肿瘤模型上CD28和4-1BB具有类似的活性,但是CD28 CAR-T表现出了更多的增殖,而4-1BB CAR-T显示有更持久的活性。第三代CARs在胞内信号结构域加上了第二个共刺激分子。因此,scFv结合TAA后激活了CD3ε第一信号和以两个共刺激分子信号为通路的第二信号。CD28结合4-1BB或CD28结合OX40作为共刺激分子在第三代的CAR-T中最为常见。截至目前,少许第三代CAR-T已经进入临床试验,其相对于2代CAR-T的优越性还需要进一步评估。理论上,只要TAA在癌细胞表面表达,CAR-T就可以识别。然而由于实体瘤表型的异质性,一些癌细胞不能被CAR-T识别,这就大大降低了CAR-T治疗的几率。第四代CAR-T,被认为是T细胞重定向,普遍的细胞因子杀伤型CAR-T细胞(TRUCKs ),用于解决由于癌细胞表型异质性带来的杀伤作用降低等问题。在该策略中,CAR-T被修饰成当CAR-T被激活时,诱导表达细胞因子如IL-12(如NF-AT(激活的T细胞核因子)控制的表达)。因此,IL-12在肿瘤细胞周围积累,引起固有免疫反应杀伤肿瘤。图4A举例说明了4代CAR-T。
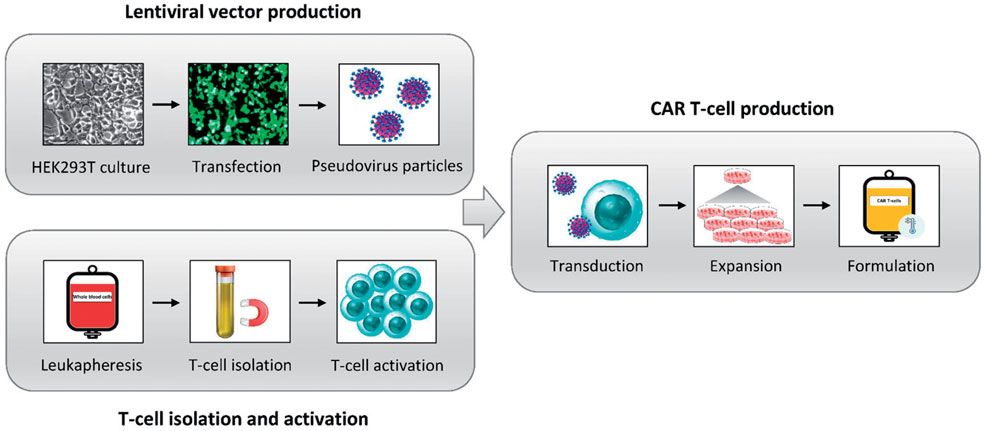
图3.应用慢病毒载体生产CAR-T的流程
针对特定TCR位置--TCRα恒定区的CARs(TRAC)
通常,基因编辑的CAR是通过病毒载体或其他载体随机整合进T细胞基因组,在这个过程中,造成了一些广谱且不适宜的整合,包括克隆增殖,基因沉默,差异性表达,以及一些有可能致癌的转化。最近,(CRISPR)/Cas9 技术已经用于CD19-CAR的构建,精确地将CAR整合进TRAC区域,这一改进,较常规的CAR-T相比,TRAC CAR-T 提高了CAR-T 的性能(图4B)。
通用的的CARs和CAR-T细胞
由于每种CAR只识别一种,最多两种靶分子的限制,有人尝试开发CAR分子可以识别任何抗原,例如将scFv替换为Avidin(生物素结合免疫受体BBIR CAR),该CAR可以结合任何生物素化的抗体。最近,还有另一种策略叫做SUPRA CAR(分离的,普遍,可编程),该策略由一个普遍的受体伴随亮氨酸拉链适配子(zip-CAR),以及一个单独的scFv伴随拉链适配器(zip-scFv)构成。该系统的优点是抗原特异性可以通过改变scFv而改变,但不影响CAR-T细胞(图4C)
目前,获准的CAR-T系统为自身CAR-T细胞。由于其昂贵的成本和时间消耗,限制了该CAR-T的治疗可行性。因为很多患者在先前的治疗中经历了化疗,淋巴细胞数目大量减少。因此,通用的CAR-T细胞治疗更有优势。更加理想的策略是通过基因工程手段消除内源性的TCR和或HLAI,以免同种异体的CAR-T引起移植免疫排斥反应。
自驱使的CARs
为了便于T细胞追踪肿瘤细胞,一些研究者在CAR-T细胞上共表达趋化因子受体,以达到与表达趋化因子的肿瘤细胞以及肿瘤相关细胞结合。例如Craddock 和他的同事借助于共表达CCR2b趋化因子受体,使anti-GD2 CAR-T细胞向分泌CCL2的神经母细胞瘤细胞归巢率提升了近10倍。类似的,在CD30-CAR-T 细胞上共表达CCR4 ,显著提升了CD30-CAR-T 向CCL-17阳性霍奇金淋巴瘤细胞的移动。(图4D)

图4.CAR-T产生和下一代CAR-T设计
武装化的CARs
通过基因工程手段修饰抵抗免疫抑制,提升治疗效果的CAR-T,称作为抗免疫抑制的CAR-T细胞或者称作武装化的CAR-T。为此,免疫抑制因子的显性阴性受体(如转化生长因子-β [TGF-β])通常与相应的CAR共同表达,以增强CAR-T细胞的疗效,尤其是实体瘤。另一种策略是开发免疫检查点疗法,招募免疫检查点抑制剂到那些广泛表达关键性抑制调节通路的肿瘤细胞和肿瘤基质细胞周围。进一步武装化的CAR-T细胞可以分泌细胞因子,或者表达可溶性配体(如IL-12)来增强细胞毒性以及抵御肿瘤微环境的作用。
On-switch CAR
也可以称作受体分离式CAR,这些CAR-T细胞设计为初始失活型,直到通过加入诱导剂诱导才被激活,完成工程化T细胞的完整信号。他们的受体包含两个独立的受体部分,一个抗原结合受体类似于传统意义上的CARs,以及胞内信号元件,二者在有小分子存在时完成组装,可以实现时间,空间上的控制以及根据CAR-T活性,在剂量上进行调节。
Off-switch CARs
也可以称作为自毁式CARs,这些细胞设计为易于清除异常细胞,包括制备进程中由于基因整合造成的的致癌转变,以及当输注的细胞毒性太强时,需要封闭掉这些毒性,就可以启动这些细胞的自毁装置而达到调节。该策略通常借助于一些自杀基因,例如疱疹病毒的胸苷激酶,可诱导的人caspase 9 ,Fas等来驱使工程化T细胞的凋亡。该策略的一个实例是在系统中运用cas9水解结构域连接在人FK506结合蛋白上,可以实现小分子雷帕霉素类似物加入,引起的条件二聚化。
Tagged CARs
标记的CAR-T细胞或杀伤开关CAR-T细胞包含一个特定的可靶向部分,该部分可被上市的单克隆抗体药物识别,并在发生不良事件时触发T细胞清除。当这些细胞的标签是肿瘤抗原时,运用单克隆抗体可介导T细胞清除,增强抗肿瘤活性。例如应用该策略的CAR-T,有表达人EGFR 多肽的CAR-T,表达CD34/CD22融合抗原表位(RQR8)的CAR-T,可以分别被cetuximab 和 rituximab 识别。如图4H
Dual CARs
二联CARs,也叫做并门CARs,表达两种类型的CAR,识别两个靶点。这些CARs的胞内结域尾部不同,一个含有CD3ε信号结构域,另一个则含有共刺激结构域。二联CARs只有在两个CARs结合他们相应的抗原时,才被激活。因此,一个抗原的结合不足以诱导T细胞反应,所以,健康组织在表达一种抗原时不会被CAR-T细胞损伤。
TanCARs
串联CARs,也称作或门CARs,在该系统中,CAR-T细胞表达双特异性CAR,包含两个scFv的串联,每个scFv识别一种靶分子,例如CD19和CD20,两个靶点都是B细胞恶性瘤的表面抗原,CD19-OR-CD20 CAR-T 可以结合一种或两种抗原来阻止CD19-细胞的逃逸。另一个实例中用HER2/CD19双特异性CAR-T细胞显示出协同的抗肿瘤活性。
iCARs
抑制型CAR(iCAR)T细胞,或称作非门CAR-T细胞,被用于需要时,限制T细胞反应。(iCAR)T 细胞表达两个独立的CARs:一个传统的激活CAR和一个抑制iCAR。T细胞的激活是通过传统的CAR与靶抗原结合实现的,而当iCAR与靶抗原结合时,CAR-T失去活性。iCAR设计保护了健康组织,免于T细胞在识别肿瘤抑制蛋白时被误伤,以及在识别那些癌细胞低表达,健康组织广泛表达的抗原时,不至于被T细胞误伤健康组织。
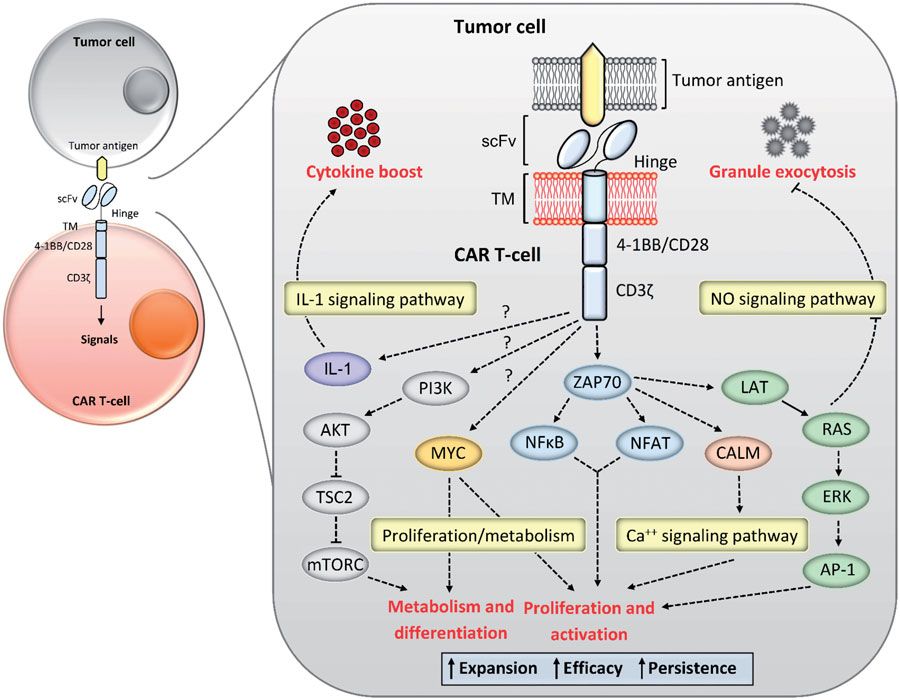
CAR-T细胞胞内级联的下游信号通路影响着CAR-T细胞在体内的有效性和持久性。对CAR - T细胞的活性和持久性所涉及的分子机制的深入理解,仍然是一个开放的研究领域。在Karlsson 和他的同事们的前沿工作中,对CAR-T下游信号通路的研究主要通过对下游多种蛋白激酶的磷酸化水平进行评估。他们强调,在三代和二代CAR-T细胞中,三代CAR-T有着本质上的高水平磷酸化和本质上的高信号强度。然而,在二代和三代CAR-T中,CD3ε, LAT, LCK, ZAP70, Paxillin,PECAM-1, RAS, ERK/MAP kinase 1/2, EGFR,和 CDK2 具有类似的磷酸化水平。CD3ε, LAT, LCK, 和ZAP70 的激活需要TCR信号,而Paxillin和 PECAM-1在大多数炎症条件下,调节T细胞粘附和白细胞跨内皮细胞的迁移。Ras的异常表达,通过抑制一氧化氮信号通路,阻止了TCR信号的级联转导,最终诱导毒性颗粒的外吐(例如:穿孔素,颗粒酶A,颗粒酶B)。
PI3K 信号通路也影响着CAR-T细胞的分化和持久性。该信号通路在CAR-T细胞中明显上调,通过mTOR介导的糖酵解驱使CD8+效应CAR-T细胞的分化。有趣的是,在体外扩增时,当抑制了抗原无关的构成型PI3K通路,可通过增强幼稚T细胞/干细胞样记忆T细胞和中央记忆T细胞群体,以及减少效应T细胞群体,从而改善体内T细胞的持久性和细胞因子的产生。然而,抗原依赖的PI3K信号与CAR-T的效能和CD8+T细胞介导的肿瘤消除有关,这就预示着CAR-T在体外扩增时抗原非依赖性PI3K激活与CAR-T体内持久性有关。此外,在一个CAR基因治疗模型中,利用组蛋白赖氨酸乙酰化的识别结构域bromodomain 和外蛋白基序的抑制剂JQ1处理CAR-T,导致体外扩增的CAR-T具有干细胞记忆T细胞和中央记忆T细胞的表型,并延长了CAR-T在体内的持久性。BET蛋白作为c-Myc信号的调节因子,可以抑制那些与Myc依赖的靶基因下调的相关蛋白质。因此,抑制PI3K和mTOR活性可诱导c-Myc信号通路,导致CAR-T细胞代谢和分化的改变。上调c-Myc,通过调节葡萄糖和氨基酸代谢促进T细胞代谢的重编程。另外,已经证实在绝大多数实验条件下,c-Myc信号通路对T细胞增殖增长是必要的。
促炎细胞因子水平(例如IFN-γ,IL-2,IL-6)的峰值水平,通常在输注CAR-T体内激活后5天内检测。IFN-γ,IL-6在CAR-T的活性,以及后续的副反应中起着很重要的作用。大多数情况下,IL-1先于IL-6产生(大约24h),推测IL-1信号通路可能在体内激活CAR -T细胞后,启动细胞因子的升高, 图5阐明肿瘤抗原与CAR结合后CAR-T细胞活化和扩增的主要信号通路。
慢病毒属于逆转录病毒家族中的一个亚科。感染颗粒为正二十面体衣壳蛋白包裹着螺旋结构的核糖核酸蛋白,衣壳被含有病毒糖蛋白的细胞脂质包膜所包围。病毒粒子包含两个RNA基因组拷贝和病毒复制酶。所有的逆转录病毒都有gag(衣壳蛋白)、pol(逆转录酶和整合酶)和env包膜蛋白基因。另外,慢病毒含有两个调节基因tat和rev,而人类免疫缺陷病毒1型(HIV-1)有额外的辅助基因,包括vif、vrp、vpu、nef,它们编码参与病毒复制的蛋白。病毒基因组还包含两个未翻译的区域5’ R-U5 和3’U3-R 末端。
在病毒附着到宿主细胞后,病毒RNA渗透进宿主细胞,并以此为模板,利用逆转录酶合成病毒cDNA,在逆转录和合成双链DNA的过程中,RNA的两端都进行了复制,产生了长末端重复序列(LTRs),在双链的病毒DNA中,每条链含有U3-R-U5序列,然后双链DNA被运送到细胞核内。新合成的逆转录病毒DNA整合到宿主DNA中,称为原病毒。原病毒在细胞周期过程中复制,然后传递给子细胞。不像其他逆转录病毒科成员,慢病毒可以有效地感染不分裂的细胞,这使它们更有吸引力用于基因治疗。最后,子代病毒基因组从整合的DNA转录到细胞质中。病毒粒子从质膜出芽获得其脂质包膜。在出芽过程中,病毒蛋白被病毒蛋白酶裂解,产生成熟的感染性病毒粒子。
慢病毒载体是最为常用的原代细胞稳定转导的基因递送系统。虽然LVs可以从其他慢病毒中获得,但今天大多数LVs来自HIV-1(图6A)。出于安全性的考虑,重组LVs设计为复制缺陷型。为了达到该目的,产生病毒所需要的元件被分割在几个质粒上。第一,编码感兴趣基因的转移质粒含有必要的顺式作用元件:(Ⅰ)侧翼的长末端重复序列LTR基因,是基因表达,逆转录和整合必要的,(Ⅱ)ψ序列是基因组RNA包装所需,(Ⅲ)逆转录反应原件RRE(可选择的),促进病毒RNA的加工和运输。第二,包装质粒,包含必要的反式作用元件:gag/pol 编码病毒复制所需的结构元件,可选择的rev用于病毒RNA转录本的核输出,tat 用于激活5’LTR弱启动子驱动的基因高水平表达。第3,包膜质粒,通常驱动病毒糖蛋白的表达VSV-G(水泡性口炎病毒的g糖蛋白),为LV颗粒提供受体结合蛋白。这样,由于包装病毒DNA所需的病毒组分与转移质粒分离,包装序列无法整合到病毒基因组中,LV无法复制。到目前为止,已经开发了三代LVs。图6(B-D):
*第一代慢病毒载体:慢病毒辅助基因vpu, vpr, vif, nef, tat, 和 rev 包含在该系统中。然而,存在一种风险是,产生病毒的宿主细胞内内源性病毒元件与重组的慢病毒递送系统的病毒元件可以互补产生有复制能力的慢病毒。因此,由于安全性问题,这些载体没有得到广泛使用。
*第二代慢病毒载体:在这个系统中,基因组元件编码的病毒辅助蛋白vif, vpu, vpr, 或nef被移除,因为它们与病毒在原代细胞和体内的繁殖有关,但对于重组LV的生产是必不可少的。因此,第二代LV系统的包装质粒编码9个HIV基因中的4个:gag、pol、tat和rev。
*第三代慢病毒载体:整合到宿主基因组的完整LTRs的存在可能存在安全风险。首先,因为在这种情况下LV转导的细胞变为被野生型病毒感染的细胞,整合的载体可以被挽救,产生重组LV不需要的复制元件。此外,LTRs的整合可以激活邻近的细胞基因,并最终激活一个编码细胞增殖相关蛋白的基因。为了克服这些潜在的风险,通过引入LTR的U3区域的缺失,开发了第三代自灭活的LVs,从而导致可能被激活的包装载体的转录失活。此外,tat从转移质粒中被删除,其转录功能通过替换U3启动子区域来完成,转移质粒中5’LTR用其他强病毒启动子如CMV或RSV启动子替换。最后,为了增加安全性,rev由一个单独的质粒编码。因此,该系统由四个质粒组成:(i)LTR部分缺失的转移载体和相关基因的外源启动子驱动表达;(ii)只含有gag和pol基因的包装质粒;(iii)rev质粒;(iv)包膜质粒(VSV-G)。
目前,CAR-T细胞主要通过病毒载体制备,大多数情况下由慢病毒或逆转录病毒做载体。与γ逆转录病毒载体相比,LVs因其更安全的整合位点而更常用于临床试验。一般来说,基于病毒的载体比传统载体具有几个潜在的优势,包括具不同病毒有不同表达特征的可用性、高转导效率、永久整合进宿主细胞基因组的能力、相对较短的转导时间,以及利用组成型生产细胞系进行大规模生产的能力。尽管有这些优点,但是基于病毒的载体仍然存在着安全隐患。LV和逆转录病毒载体的半随机和不受控制的整合进宿主基因组导致插入突变,产生一定程度上的工程化细胞具有癌变的潜在风险。此外,可变的拷贝数可以通过病毒载体转移并在T细胞表面表达,这可能导致异质性的CAR-T细胞群,具有不同的细胞毒性能力。为了克服这一局限性,重要的基础和临床研究致力于开发替代的非病毒载体系统,如微圆环DNA、转座子、位点特异性编辑工具、CRISPR/Cas9技术和分子偶联。最近,CAR-T细胞已经通过转座子成功构建,包括睡美人转座子,转座因子系统。这些系统具有比病毒载体更大的转基因能力。然而,由于它们随机插入的特点,仍然需要考虑其临床应用的安全性和有效性。位点特异性编辑工具如TALENs和ZFNs可以用来解决基因组特定基因座整合的问题。为了使它们在临床试验中更受欢迎,需要降低设计和优化每个目标位点的酶的成本,提高它们的效率。新兴的CRISPR/Cas9技术正在努力产生高度均质化的CAR-T细胞群。使用位点特异性编辑工具,通过特定位点的整合,产生内源性TCR被剔除的CAR替代的TRAC CAR-T细胞。虽然这项技术为研究人员提供了精确制造T细胞的宝贵经验,但它的编辑效率与病毒转染的编辑效率并不匹配。此外,利用CRISPR/Cas9进行相对较大的转基因转移仍然是一个挑战。考虑到所有这些要点,非病毒载体有一些明显的局限性,它们的效率仍然无法与病毒载体相比。此外,在考虑在临床试验中使用这些方法时,还应解决其他问题,包括体内性能差、生理环境不稳定和细胞摄取障碍。
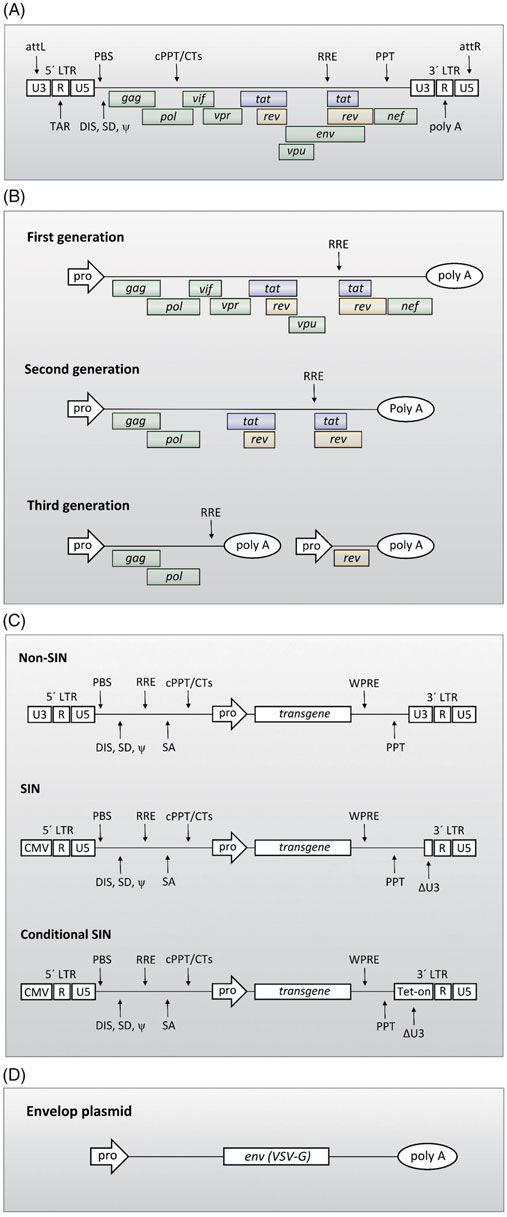
图6.来自于HIV-1慢病毒载体的进化历程
瞬时和稳定转染
为了产生足够数量的基因工程化T细胞用于临床实验,生产大量的慢病毒是必要的。通常,慢病毒通过瞬时转染HEK293(T)细胞制得。值得注意的是,为了根据GMP生产LVs,生产中使用的细胞系应符合细胞基质指南(如CPMP /ICH/ 294/95指南)。使用磷酸钙、聚乙烯亚胺等阳离子试剂或脂质体(lipofectamineVR、fugeneVR293fectaminVR)进行转染。尽管用磷酸钙和阳离子脂类进行小规模转染可以获得相当高的转染率,但这些方法要么难以规模化,要么成本非常昂贵。基于稳定包装细胞系的LVs的制造已经发展到临床应用。虽然慢病毒的瞬时转染更快更有效,稳定转染的细胞系产生的LVs更适合临床应用。由于其整体安全性和可重复性,稳定的包装方法使大规模生产的时间和成本降低。不幸的是,由于gag、pol、rev和VSV-G的细胞毒性和细胞抑制作用,这一策略变得很复杂。包括“tet-on”和“tet-off”在内的诱导表达系统已经被应用来克服这个问题,这些基因的表达分别通过在细胞培养基中添加或去除四环素来调节。
贴壁与悬浮培养
对于小规模研究用LVs,贴壁细胞制备就足够了。使用贴壁细胞系产生更大的病毒剂量是有限的,因为需要一个表面面积很大的的细胞培养容器培养细胞。贴壁培养可以通过增加培养单元来达到增加培养面积。培养皿,T型烧瓶,多层系统(例如细胞工厂),滚轴瓶也是大规模生产的培养系统。这些培养方法,同时需要多个培养箱放置,这本身就需要很大的培养空间。Kutner 等人观察到用于LV生产的细胞系在HYPER Flask中生长速度比传统培养方法快10倍左右,从而产生更高的单位面积的病毒滴度。然而,与其他系统相比,在载体滴度方面没有发现明显的差异。
最近,一种基于中空纤维生物反应器的新技术被引入到贴壁细胞培养环境中,用于大规模的LVs生产。中空纤维生物反应器是一种全自动的封闭系统,它有成千上万的多孔毛细血管(中空纤维),细胞在纤维周围的毛细血管外培养。这种生物反应器的局限性是需要建立多个并行系统才能实现大规模生产。
工业生产慢病毒的另一种策略是让包装病毒所需的宿主细胞从贴壁培养向适应悬浮培养驯化。悬浮培养具有在搅拌生物反应器等系统中大规模扩增产生lv的细胞的优点。与贴壁细胞培养相比,悬浮适应细胞可以在大体积,小空间的环境下生产LVs,因为它们可以在不需要粘附表面积的情况下进行高密度扩增。此外,悬浮培养的另一个优点是,可在无血清的情况下生产供临床使用的LVs,从而减少外来因素的潜在污染。
各种容器培养系统已被开发,以增加细胞生长在悬浮液,包括摇瓶,波袋,搅拌罐生物反应器,玻璃生物反应器,不锈钢生物反应器。另外,也有许多研究报道了成功使用生物反应器,驯化HEK293悬浮培养产生高滴度的LVs。有趣的是,在悬浮培养法中,使用聚乙烯亚胺对HEK293T细胞进行瞬时转染,用于LV的生产,在一次性生物反应器中进行了50 L规模的生产。另一个系统是iCELLisVR纳米系统(Pall, NY),它被用于在由聚酯微纤维大分子载体组成的固定床生物反应器中,通过瞬间转染贴壁的HEK293T细胞来产生LVs。该系统可在灌注培养下进行,接近6L的培养系统中产生滴度高达2x106 TU/ml(转导单位)的LVs。iCELLis生物反应器通过减少接种量、降低培养pH值、测量细胞代谢来控制灌注率,从而优化LVs的生产。
感染复数
影响转导效率的一个关键参数是感染复数(MOI),MOI指的是每个细胞感染的病毒粒子个数,病毒粒子滴度以感染单位(IU)/mL 或 TU/mL表示 。LVs的感染能力随细胞类型的不同而不同,因此每种不同的细胞类型需要不同的MOI。理想情况下,MOI与感染细胞数量之间应呈线性关系。为了确定T细胞有效转导所需的最佳MOI,可以使用一个表达报告基因如绿色荧光蛋白的LV,测试一系列MOI(如MOI)。然而,当考虑到患者的特异性变化时,用于描述转导条件的MOI的优化就变得更加困难。
在LVs生产的下游过程中有三个不同的阶段:(i)捕获阶段,它消除污染物,包括添加的材料、血清蛋白和质粒DNA(在瞬时转染中),以及来源于宿主细胞的成分,细胞碎片,宿主细胞蛋白,宿主细胞DNA片段,产生澄清的细胞粗培养物。ii)中间纯化阶段,通常伴有25-50 U/mL核酸酶处理,在澄清的上清中添加核酸酶,进一步去除特定的细胞,病毒或加工过程中产生的杂质(如蛋白、DNA、质粒残留和内毒素)(iii)抛光阶段,这是在合适的配方缓冲液中去除痕量污染物和小杂质以获得GMP级产品的最后一步。
捕获
低速离心和微滤用于实验室和大规模捕获过程。尽管微滤的效率很高,然而随着时间的推移,细胞碎片堵塞微孔造成病毒颗粒截留是制约微滤的最大屏障,限制了LV的回收率。因此,大多数已发表的研究都集中在深度过滤的使用上,深度过滤已被证明可以最大限度地减少过滤器堵塞和在澄清步骤中病毒颗粒的损失。深度过滤通过一系列孔径减小的膜来实现,一般从0.8-0.45μm该方法用来澄清多种包膜(如LVs),非包膜病毒颗粒(腺病毒),病毒回收率达到90%。深度过滤的高效率是通过深度相关的尺寸分离和最先进的深度过滤膜的带电性质及其三维结构来实现的。另外,需要格外关注过滤过程中的流速(ml/min)和剪切力(S-1)。例如,有研究表明较高的流速(如100 mL/min比50 mL/min)可导致更高的LV得率。然而,微滤的流量需要根据过滤类型进行优化。剪切力5000 S-1,,流量10 LMH(L/m2/h)是已经经过优化的微滤参数。高剪切力可能影响LV的结构完整性,减少LV的回收率。
LV颗粒的浓缩是澄清过程下游的一个常见步骤。其目的是减少进料量,从而减少对下游设备和材料的投资。虽然较高的浓度可以导致较低的生产成本和杂质,值得考虑的是病毒粒子过高容易产生聚集,例如一些病毒粒子在很高浓度下由于表面的相互作用发生聚集。超滤和超速离心(UC)是LV浓缩最为常用的方法。与大规模生产一致,更多的人喜欢用超滤的方法浓缩,比起超离,超滤有更易于放大和产生更高滴度的功能性病毒载体的优点。另外,超离在大规模生产中更耗时,由于超速离心机的容量有限,多次离心,需要大量的时间。此外,在gmp实验室设施中还没有常规的超速离心。切向流过滤,截流量为750 kDa(~50nm ),是一种代替超离的方法,特别适用于LV颗粒悬浮液的浓缩和随后的过滤,使之进入最终的配方缓冲液。与传统的直接流过滤不同,切向流过滤在孔径从100到500 kDa不等的膜表面上切向通过LV上清液,因此减少了过滤器的污染和LV的滞留。该方法可以有效浓缩原始LV粗提上清,可实现2000倍的浓缩,且回收率高达97% 。
中间纯化
色谱法由于其高回收率、可扩展性和一致性而成为大多数大型LVs制造公司的首选方法。在多种色谱法中,阴离子交换色谱(AEXc)被认为是最适合大规模纯化的方法。然而,由于LVs的膜成分不确定,AEXc树脂的选择仍然很复杂。对于固定相,整体吸附剂被越来越多地使用,因为它们互联的大通道提供了高流速和物料迁移。高流速的单片填充床的相容性使其纯化时间缩短,保持了LV的高感染率。此外,由于其较高的床层孔隙率,整体支架与颗粒基树脂相比,可产生更低的压降,从而实现了高效的LVs纯化过程。Bandeira 等人研究结果表明CIM® 是一款DEAE 阴离子交换整体柱,具有高度互联的甲基丙烯酸酯聚合物通道网络特征,与膜吸附相比,有较高的回收率,洗脱回收率为80±5%。CIM® DEAE整体柱有效的纯化,通过0.65 mol/L NaCl 梯度洗脱得到。与之相对的是,使用其他阴离子交换膜(例如Q膜层析)需要浓度在1-1.5mol/L NaCl 洗脱多达四个馏分。这是一个非常关键的参数,因为LVs对高浓度的盐非常敏感,之前有报道称将LVs分别置于高浓度的盐缓冲液中1和2h,结果病毒的感染力分别下降了16.0±6.4%和22.1±6.4% 。CIM® DEAE整体柱明显的优于传统的树脂(载量,样品分散度)和膜系统(即对流质输运、低压降、高流量),导致高分辨率的分离,这可以归因于空隙体积的缺失,从而减少“涡流色散”。然而,需要注意的是,CIM® DEAE整体柱对LV颗粒的高回收率明显依赖于分步洗脱类型,当线性盐梯度洗脱时,回收率低至65%。
抛光和制剂配方
在抛光阶段,微量的污染物和杂质,通常被称为“异构体”,和可疑的泄漏产品被清除。然而,抛光和随后的0.2μm过滤除菌通常导致终产物的大量损失和稀释。由于其高效性和稳定性,大规模纯化,典型的使用分子筛作为最后的抛光步骤。这种方法对于去除比基质孔径小的污染物(如盐、糖、小分子)非常有效。进一步的,LV上清的蛋白质污染可以通过分子筛快速高效的清除。色谱基质孔径的阈值设置得足够低(750 kDa,相当于50 nm),以防止80-120 nm的LV颗粒滞留。
在临床试验中,LV悬浮液被制成液体,储存在≤-70°,冷冻条件下运输。将LV颗粒制成培养基制剂在转导T细胞过程中是很方便的。为此,商业化的培养基包括X-VIVOTM(Lonza,Basel, Switzerland)和CellGro ®(CellGenix, Freiburg,Germany)被广泛使用。Ausubel 等人使用磷酸盐缓冲液包含4 g/100 mL 的乳糖作为LV制剂。这些一般制剂缓冲液兼容客户使用其他不同的培养基进行转导。Deb 和他的同事比较了一些水溶制剂缓冲液,建立了稳定慢病毒制剂的平台。他们发现与传统的HEPES LV制剂相比,包含20 mmol/L 1,4-哌嗪二乙磺酸(PIPES) ,75 mmol/LNaCl,2.5%蔗糖(w/v),PH6.5的缓冲液中,LV具有很高的稳定性和很好的生物学特性(例如很好的抵抗热聚集,耐高盐离子浓度,多次反复冻融后感染力损失不大)。有趣的是,基于PIPES 的缓冲液,LVs对原代T细胞的转导能力比基于HEPES和组氨酸缓冲液中的LV的转导力分别高出25%和35%。尽管如此,考虑到LV生产加工过程中的众多挑战,选择适宜的缓冲液仍然存在争议。
LVs的滴度测定
LV产品在临床试验中的应用要求准确调整载体剂量和细胞转导效率(转导效价)。标准方法需要测定LVs IU/mL或TU/ mL。这是一个功能性效价,表明了LVs在特定条件下转导特定细胞系的能力。理想情况下,应该使用最低水平的慢病毒转导与足够的基因表达。病毒的效价,尤其是LVs,依赖于严格的测定方法,载体类型(二代,三代或反式慢病毒系统),以及用于测定效价细胞的条件。
对载体效价的测定一般分为功能性和非功能性的(有货没有荧光标签),或者采用物理学或生物学的测定方法。物理学或非功能学的测定方法基于对载体颗粒组成数量上的评估(例如P24衣壳蛋白的浓度/RT 活性/基因组拷贝数目),生物学或者功能上的评估主要计算感染细胞中前病毒载体拷贝数目或者报告基因或转导基因在感染细胞中的表达情况。
通常物理方法高估了载体功能效价,在预测载体转导效率方面价值有限,因为在此过程中,计算了所有的感染性和非感染性病毒粒子。例如,有一种方法是通过P24 ELISA测定,得出LV病毒载体上P24衣壳蛋白的浓度。该方法可以测定总的病毒粒子浓度(物理效价),方便检测病毒载体生产包装的效率。大多数试剂盒通过ELISA定量LVs上清中总的p24抗原。因此,该技术可以定量游离的和非功能性载体粒子表面的P24。然而,一些试剂盒像QuickTiter Lentivirus Titer ELISA Kit(CellBiolabs, San Diego, CA),使用独特的试剂分离慢病毒粒子去除了游离的p24,因此,该试剂盒只定量了存在病毒粒子表面的p24。
目前,通常存在两种功能性效价测定方法:(1)利用q-PCR的方法定量载体DNA的拷贝数目,(2)定量转导细胞中转基因的表达。通过转基因表达检测转导细胞,是测定感染性LV滴度最可靠的方法。该方法仅可用于携带可靠标记基因的载体的效价测定,这些标记基因可通过显微镜或流式细胞术进行监测,或在有针对LV编码蛋白的特异性抗体时进行检测。在这些检测中使用的标记基因包括neo抗性基因、绿色荧光蛋白或其他活性颜色。所使用的标记基因应由在转导细胞中活跃的启动子控制(如HEK293T)。然而,局限于报告蛋白的转基因表达功能分析不能区分单个或多个整合事件的细胞,而且在组织特异性启动子的控制下测量转基因表达可能不可靠。借助于q-PCR检测前病毒DNA在感染细胞中的拷贝数目来定量感染载体的拷贝数目。这是测量慢病毒滴度最准确和最直接的方法。q-PCR技术独立于启动子功能,允许转导事件的绝对定量。然而,对于载体功能性测定的最佳方法是对基于启动子驱动的转基因产物的测定。
二者结合起来,是目前广泛接受使用的标准方法,方法的选择很大程度上依赖于实验设置。一种理想的载体滴度测定方法应该同时兼容多种载体滴度测定,而且尽可能的减少操作时间。考虑到现有方法的局限性,建议使用双滴定-荧光激活细胞分选仪和q-PCR,同时使用绿色荧光蛋白载体和其他载体,并对两种方法的结果进行比较。使用q-PCR和流式细胞术计算LV的TU/mL常用公式如下
Titer(TU/mL)=(NXCorFXD)/N
在这里N代表第一天里每个孔细胞数目的平均值,C(q-PCR)是每个二倍体基因组中慢病毒载体的拷贝数目,F(流式细胞术)荧光细胞百分比,D 是载体储存浓度的稀释倍数,V是载体的稀释体积。
GMP是一种用于制药工业的质量保证系统,以确保最终产品符合预定的规格。这些指导方针适用于生产过程的所有部分(原材料、生产空间、设备、人员、文档、批到批一致性等),实现生产质量一致的产品,并尽量减少污染。关于GMP级别的基因和细胞治疗,一些指令和指导方针已由几个国际机构定义,包括美国药典(USP:1043,1047),欧洲药典(EP;0784,2034,0153),食品药物管理局(62,FR 9721,21 CFR 1271),欧洲药物机构(EMA; EMA/37318/2018, EMEA/CPMP/BWP/2458/03, CPMP/BWP/3088/99),和国际协调委员会(ICH; e.g. document ICH S6)。在这些指南中,EMEA/CPMP/BWP/2458/03指南描述了应用于体外和体内非临床试验的LVs的一般质量要求。因为不可能讨论LVs整个生产过程的所有GMP条件(即原材料、设备、人员等),在这篇综述中,我们将重点介绍LVs生产过程中与产品和流程相关的参数。如今,针对每一种病毒载体,已经开发和优化了大量的QC检测方法,而且在任何拟供人类使用的产品发布之前,必须满足获得GMP认证的标准。为了进入临床试验,需要对LVs的GMP生产进行一些特性测试,包括评估载体的纯度/特性(例如,检测载体的纯度/特性)。蛋白质和DNA污染物、LVs形态和基因组完整性、安全性(如缺乏复制能力强的慢病毒、外源性病原体,支原体,内毒素/热原,效力(总/活性LV粒子),以及最终批次的其他理化性质(如pH、渗透压、电导率)。然而,由于LV在体内外基因治疗方面的应用经验有限,加之LV颗粒的复杂性,所以针对LV的QC规定的指导方针非常宽泛,数量有限。此外,建立完全的GMP生产,需要大量的投入,因此,绝大多数制造商更喜欢从临床前阶段向生物许可证申请和商业化生产阶段的进展过程中提高GMP标准水平。根据多个检测变量(如靶细胞、接种量、孵育时间)的影响,不同实验室获得的LVs制造的QC结果几乎没有可比性。因此,需要为不同的QC参数提供一个特别的标准化平台,从而获得相应的监管批准。表1总结了GMP条件下LVs生产评估的法规要求和规范。
分离富集T细胞
Ficoll密度梯度离心法是去除红细胞和血小板的常用方法。此外,一些自动细胞收集器如COBE 2991 Cell Processor(Terumo BCT,Lakewood, CO), Haemonetics CellSaver(Haemonetics Corp., Braintree, MA), Baxter Cytomate(BaxterOncology, Chicago, IL), Biosafe Sepax II(GE Healthcare Life Sciences, Marlborough, MA),和 CaridianBCT Elutra(CaridianBCT, Lakewood, CO)用于T细胞的分离。为了去除特定的细胞群(如单核细胞,NK细胞群),经常使用磁珠分选系统。此前大量试验证实,大量的未知细胞阻碍T细胞的存活。Milteny开发了一种全自动的封闭系统CliniMACS系统(Miltenyi Biotec, Bergisch Gladbach,Germany),将各自独特的抗体偶联到磁珠上用来从异质性细胞群中分离纯化不同的细胞群。
分离完成后,细胞通常需要冷藏保存在血袋中,冷链运输到中央工厂后进行一些列富集,激活,转导步骤。为了减少冷冻和解冻带来的基因上的一些改变,已经提出了一些建议。T细胞分离是分离外周单核细胞关键的一步,以便起始培养细胞以高纯度的细胞培养开始,特别是在急性淋巴性白血病患者中存在高浓度的恶性循环B细胞。截止目前,在CAR-T生产中,已有anti-CD3+(T-细胞), anti-CD4+(辅助T细胞), anti-CD8+(效应T细胞), and anti-CD62L+(中央记忆T细胞)的磁珠系统用于富集T细胞。
激活的T细胞
为了避免在体外T细胞激活步骤中使用抗原呈递细胞的繁琐过程,已经建立了几种方法:(1) 单克隆抗体和白介素刺激,如anti-CD3 和CD28mAbs, OKT3(anti-CD3 antibody), IL-2, IL-7, 和IL-15,诱导产生了高比例的记忆细胞亚群。(2)细胞大小的磁珠微球,磁珠微球用CD3,CD28抗体包被,可强烈地诱导记忆和效应T细胞的增殖,细胞因子的分泌量提高了10-100倍,甚至比使用抗CD3抗体和IL-2刺激效果还好。(3)人工抗原递呈细胞如K562细胞,因为它可以激活T细胞,CAR-T细胞,主要是由于K562不表达HLA A和B,避免了刺激同种异体T细胞,而其表达内源性的共刺激分子(如CD40, CD40L, CD70, CD80, CD83,CD86, CD137L, ICOSL, GITRL, 和CD134L)可以方便地转导表达TAA(如CD19)
T细胞转导
LV转导是临床试验中第二大常用的基因传递方法。然而,一些基于质粒的基因传递系统,如转座子/转座酶系统(如SB和piggyBac)也有人提了出来。尽管利用SB系统来传递和整合目标基因的效率已经接近病毒转导的效率,不足之处仍然存在。例如,制备CAR-T需要的时间远远高于病毒系统。因此,最近一项基于病毒载体的CAR-T细胞的研究表明,通过将扩增时间从9-14天缩短至3-5天,可以大大缩短CAR-T细胞的产生时间。有趣的是,与后期(第9天)收获的CAR-T相比,早期(第3或5天)收获的CAR-T显示分化较低,体外抗白血病的活性增强。
SB基因整合系统可能会有效降低靶细胞插入突变产生的致瘤性。相比之下,PiggyBac系统有几个胜过SB系统的优点,包括高转到效率,转移大片段DNA的能力,可以插入高达几十万个碱基。
CAR-T扩增
在临床应用中,CAR-T细胞的扩增需要将培养物从培养皿/培养瓶转移到生物反应器中。已有许多生物反应器平台,例如GE WAVE生物反应器系统(GE Healthcare LifeSciences, Marlborough, MA), G-Rex 生物反应器(WilsonWolf, New Brighton, MN), CliniMACS系统等可便捷的进行CAR-T的扩增。如前所述,抗CD3和CD28微球被广泛用于实现CAR-T细胞的持续激活和扩增。然而,这些微球倾向于聚集和沉淀与系统底部,使细胞分离过程增加难度并延迟。这一问题导致了CAR-T细胞功能减弱和最终产物的损失。CD3/CD28的司具体抗体复合物(ExpamerTM, Juno Therapeutics, Seattle, WA)已经被开发了出来用于解决局上面微球的问题。CAT细胞扩增需要重点考虑的问题是如何保持CD4+和CD8+T细胞的平衡。而且,非转导的T细胞在有外源刺激因子刺激时也可以扩增。因此,基因工程化修饰的细胞(K562)被用做人工抗原递呈细胞来刺激CAR-T细胞特异性扩增。这些细胞都是符合GMP标准,基因工程化修饰表达一系列共刺激分子便于激活CAR-T细胞和扩增。
CAR-T细胞制剂
CAR-T细胞的配方仍然是一个很大的挑战,需要考虑很多关键因素,例如CAR-T细胞的维持条件,保存时间,在冻存和解冻过程中如何保持其活性和功能。低温储运是广泛应用的方法,在CAR-T配方中相对容易建立的操作。据研究报道,该方法不会损坏CAR-T细胞的分化,功能,和运动,而且低温保存10个月的CAR-T,再次解冻,细胞存活率仍保持在90%以上。最近,一项CAR-T低温保存的研究结果可以在实验室和大规模筛选中应用,其组分是生理盐水中包含8%的人血清白蛋白和15%的DMSO(DMSO终浓度为7.5%)或CryoStor10(BioLife Solutions, Bothell, WA)培养基中含10%DMSO(DMSO终浓度 5%)。两种冻存液尽管有相似的存活率,但CryoStor10显示出更好的复苏效果,意味着该冻存液有着比生理盐水更好的储存效果。需要进一步的评估证实该发现。有关CAR-T细胞配方的研究有限,需要进一步的开展相关研究。
CAR-T在血液癌症和非血液癌症方面的临床试验
有关CAR-T用于临床试验最早的报道是10年以前,自那时起,CAR-T细胞已经成为针对淋巴瘤和白血病中cd19阳性肿瘤细胞的一种有效且成功的治疗方式,并且该领域正在向实体肿瘤扩展。截止2018年10月,已有400多项临床试验将CAR-T细胞疗法应用于各种癌症。大多数CAR-T临床试验在亚洲进行,特别是中国(~55%临床试验),这是目前CAR-T临床研究领域最活跃的区域。北美和欧洲的CAR-T临床试验分别为33%和10%。图7A展示了目前全球范围内CAR-T临床研究的区域分布。在血液学和非血液学恶性肿瘤中,CAR-T细胞治疗的新靶点的研究进展令人印象深刻。针对18种肿瘤表面抗原的CAR-T细胞治疗已进入临床试验。
此外,许多靶抗原,由于其在癌细胞中的特异性表达,正在进行临床前研究。除了CD19,还有其他几种抗原,如CD5、CD20、CD22、CD30、CD123、CD33、CD38、Ig k、ROR1、EpCam和BCMA已被考虑用于CAR-T细胞治疗以靶向血液恶性肿瘤。有趣的是,随着CAR-T细胞治疗的扩大,LVs在CAR-T细胞的产生过程中起着关键作用。因此,在2014年,大约54%的人使用了LVs CAR-T细胞治疗试验。图7B
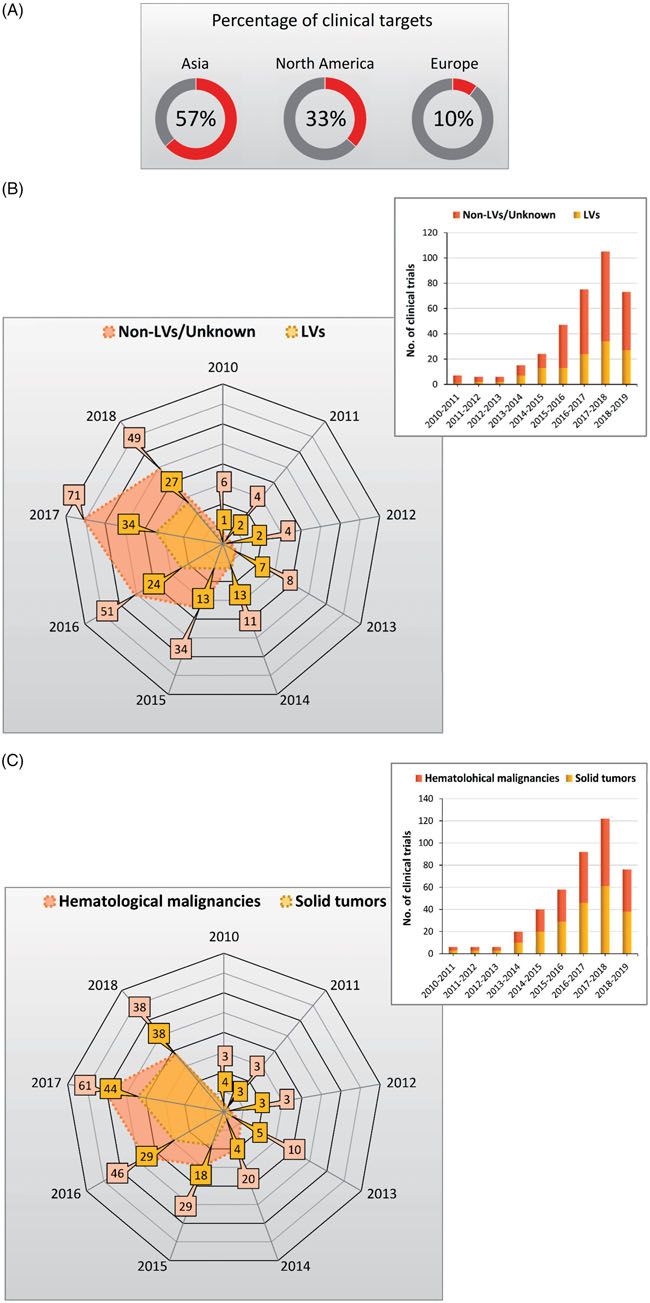
图7. CAR-T基因治疗临床试验
CAR-T细胞治疗血液病恶性肿瘤的疗效已被证实,但在实体肿瘤中,可能由于肿瘤细胞的异种抗原表达和肿瘤微环境的免疫抑制作用,其治疗效果一直存在争议。因此,使用CAR-T细胞治疗实体肿瘤的临床试验数量低于血液系统恶性肿瘤(图7C)。这就促进了更多的尝试,鉴定新的靶点用于CAR-T靶向实体瘤。例如Her2,FAP, MSLN, GD2, CD70, MUC1, GPC3, IL13Ra2, PSCA,EGFR, VEGFR2, CEA, MET, PD-L1, PSMA, ROR1, EpCAM,和 EphA2, cancer/testis antigens, GUCY2C, TAG-72,和 HPRT1 已经被评估用于临床试验。在实体瘤中,CAR-T治疗常伴随一些致命的并发症,尤其是细胞因子综合征(CRS)和CAR-T相关的脑病综合征(CRES).因此,在转移性肾细胞癌患者的一期试验中,第一代CAR靶向CAIX的T细胞表达遇到了意外的肝毒性,这使得一些患者不得不停止治疗。所以,需要控制CAR-T细胞在体内的扩增和活性。
副反应
细胞因子综合征
CAR-T细胞疗法可以引发一系列与传统疗法不同的副作用,迄今为止,CRS是最普遍的不良后果。CRS是由于免疫激活的开始导致炎症细胞因子水平升高,反过来就是治疗有效的信号。细胞因子水平明显提升的有IFN-γ,GMCSF, IL-1,IL-6和IL-10,已经在CAR-T治疗过程中报道过。虽然细胞因子是杀死肿瘤细胞所必需的,但它们也可能引起显著的副作用。因此,CAR-T细胞疗法是一把“双刃剑”,它会导致大量细胞因子进入血液,并可能危及生命。比较研究中出现的CRS严重程度和结果,一些CRS的分级标准被制定了出来,来统一评估CRS,CRS发作,CRS消退。此外,预测一些病理生理和分子生物标志物,如发热和MCP-1的结合可能有助于临床医生进行干预,并可能预防或减轻CRS和/或神经毒性;需要进一步的临床研究来证实这些预测生物标志物。
在少数情况下,CRS可以持续到2周后,病人可能需要转到ICU用于血流动力学稳定。实时监测血清细胞因子以识别CRS进程,面临着技术上的困难和高昂的成本。最近,肝细胞对IL-6产生反应的c反应蛋白被用作CRS发生和严重程度的临床标记。据报道,复发性/难治性B细胞患者的CRS发生率为19 ~ 43%。这种差异可能与该综合征的临床识别、嵌合受体设计和注入的细胞表型有关。
CAR-T细胞的毒性
多种CAR-T相关的毒性基本上包括“on-target on-tumor”, “on-target off-tumor” 和神经毒性。最常见的毒性是“on-target on-tumor” 主要由细胞因子释放过多或肿瘤细胞大量死亡引起的CRS和肿瘤裂解综合征。“On-target off-tumor” 指的是CAR-T细胞对正常组织的攻击,是最为显著的毒性。肿瘤免疫治疗最关键的问题是选择理想的靶点,该靶点明确的表达于所有肿瘤细胞,最好是为肿瘤细胞存活提供关键信号的分子。然而,由于大多数CAR-T识别靶点在正常组织中有表达,不同程度的“On-target off-tumor” 毒性是可预期的,从易控制的细胞系清除到严重的毒性,这些都是由于CAR-T细胞攻击正常组织造成的。CD19特异性CAR-T以B细胞白血病和B细胞淋巴瘤细胞表面CD19为靶点。这一抗原也在正常B细胞表面有表达,因此CD19 CAR-T“on-target off-tumor ”正常细胞,造成B细胞发育不全和低丙球蛋白血症使得患者易于感染甚至威胁生命。病人的免疫球蛋白G水平可以通过静脉注射免疫球蛋白来提高。同样的,其它一些“on-target off-tumor ”毒性,HER-2/neu特异性CAR-T细胞可引起呼吸衰竭、多器官功能障碍,并可对表达HER-2/neu的正常肺组织产生反应而死亡。
神经毒性,或CAR - T细胞相关脑病综合征,典型表现为神志不清、谵妄、表达性失语、一定程度的肌阵挛,偶尔伴有脑水肿和癫痫。这种不良结果,即CAR-T 治疗的另一个可能的副作用,在大多数情况下是可逆的。虽然神经副作用的确切病理生理学还不清楚,但人们认为神经毒性是细胞因子水平升高的结果。此外,一些研究表明,尽管在受影响的脑区缺乏CD19表达,但神经毒性与脑脊液中CAR-T细胞的存在存在一定的相关性。然而,所有神经毒性患者的脊髓液中均未检测到CAR-T细胞。神经毒性的病理生理学,以及它是来自于CAR-T细胞对中枢神经系统组织的直接攻击,还是来自于广泛的细胞因子介导的炎症,还没有得到研究。
缺少持久性
一些因素影响着CAR-T在体内的持久性:病人的前期治疗,起始CAR-T的质量和表型,培养条件和只被极少数,基因整合的方式,最终产品中CAR的分子设计,宿主对输注细胞免疫反应。此外,已证实非衰老的,非耗竭的T细胞有明显的抗肿瘤活性目前,研究正在进行关于解决CAR-T复制和寿命有限的问题,以增强T细胞在体内的持久性。例如,当修饰端粒酶的逆转录酶mRNA瞬时传递到CD19CAR-T中,研究表明,观察到这些细胞在B细胞恶性肿瘤小鼠移植瘤模型中的持久性和抗肿瘤作用得到改善。此外,其他研究表明,淋巴衰竭有利于增强体内CAR-T细胞的持久性。选择合适的T细胞表型另一个影响CAR-T细胞在体内持久性的关键因素。拥有中央T细胞表型(CD62L+)和/或记忆干细胞表型(CD45RA+)的CAR-T细胞表现出更强的体内扩增活性。另外,共刺激分子结构域如4-1BB, OX40(TNFRSF4), 和 ICOS 也可以影响CAR-T细胞持久性。
批间效应
在实体肿瘤中,肿瘤微环境是CAR - T细胞的严重屏障,在缺氧、营养缺乏、必需氨基酸缺乏、肿瘤微环境中代谢酸浓度高的情况下,CAR - T细胞可能无法增殖。此外,肿瘤组织中可能存在调节性T细胞、肿瘤相关巨噬细胞和肿瘤相关中性粒细胞等多种免疫抑制细胞。这些细胞产生多种免疫抑制因子和细胞因子,如TGF-β、PGE2、IL-10、IL4,以及活性氧,有利于肿瘤存活,抑制细胞毒性T淋巴细胞反应。为了克服这些瘤内免疫调节活动,Bollard 等人设计产生了表达TGF-β受体负调控结构域的CAR-T,以抵御内源性TGF-β产生的抗增殖和抗细胞毒性效应。最近,CAR-T细胞被改造成分泌scFv对抗免疫检查点,如PD-1。该策略成功阻断了PD-1与PD-L1的结合,提高了CAR-T细胞治疗在体内的整体抗肿瘤效果。为了改善CAR-T细胞向肿瘤的转运和浸润,提出了几种策略,包括使用特定趋化因子受体、整合素或选择素和免疫检查点抑制的T细胞转导。此外,局部注射CAR-T可以促进T细胞与癌细胞的相互作用。正在进行的临床试验将有助于评估CAR-T细胞定点给药的效率。虽然CAR-T细胞的区域传递可能弥补了t细胞转运的不足,但是局部注射CAR-T细胞比静脉注射更具挑战。同时,配合局部灌注,数量较多CAR-T细胞会聚集在肿瘤的特定位置,但是他们迁移到其他肿瘤区域的能力仍有争议。在另一项研究中,表达GD2-CART和CCL5的溶瘤腺病毒载体增加了CAR-T细胞的浸润,限制了神经母细胞瘤的进展。
自体CAR - t细胞在肿瘤的免疫治疗中显示出了巨大的优势,为个性化治疗开辟了新的领域。多种策略已应用于GMP级CAR-T细胞的生产,以病毒为载体的方法占主导地位。在病毒载体中,LVs以其独特的优势广泛用于基因治疗,转导分裂和费分裂细胞,高度整合进宿主基因组,很好的的大片段整合效率,延长的转基因表达。因此,成功的工业生产级载体最重要的一步是产生足够数量的CAR-T细胞用于输注。然而,这些病毒载体有一些缺点,包括可能的插入突变,在包装构造中调控蛋白的存在(tat, rev),整合缺陷载体的瞬时表达,以及对pH变化和高盐浓度的敏感性,这限制了它们在临床研究中的应用。因此,基于非病毒载体的下一代自体CAR-T细胞(即SB和piggyBac转座子)也被引入临床应用。尽管几乎所有CAR-T细胞研究,目前使用的是自体t细胞,这些疗法存在一些局限性,包括成本、收获和制造过程中的不确定性、产品的可变性和QC、自体CAR-T细胞生产过程中的疾病进展、肿瘤细胞污染和T细胞功能障碍。因此,目前利用新技术设计和制造同种异体CAR-T细胞的研究应在该领域取得重大进展。现成的同种异体CAR-T细胞有可能为无法等待长时间生产过程的高危患者提供一种安全的CAR-T细胞替代来源。健康的供体异基因CAR-T细胞可以从hla匹配的造血干细胞移植供体中获得。此外,基因编辑方法(CRISPR/Cas9、ZFNs、TALENs、megaTAL核酸酶和工程I-CreI归巢内切酶)可用于生产非hla匹配患者的同种异体CAR-T细胞。然而,尽管基因编辑的同种异体有较好的抗肿瘤效果,但是一些CAR-T面临意想不到的风险,例如当切割发生在多个位点时,引起的基因的脱靶分裂和不必要的易位。尽管如此,“现成的”病毒特异性CAR-T细胞也可作为一类部分HLA匹配(1-4等位基因)的t细胞使用,这些t细胞在出现反应的患者中表现出最小的GVHD,尽管它们的效率很低。这些细胞也是制造同种异体CAR-T细胞的一个有吸引力的来源。近年来,诱导多能干细胞CAR-T细胞和TRAC CAR已经成为CAR-T细胞的来源。然而,这些异体CAR-T细胞的安全性和有效性尚不清楚。值得注意的是,制造同种异体CAR-Tcells的不同方法可能会在不同的给药时间点出现GVHD和/或排斥注入细胞,因此需要相对较长的重症监护随访期。淋巴耗损性化疗和免疫抑制的宿主细胞如调节性T细胞的减少,可能阻止GVHD和同种异体CAR-T细胞的排斥反应。此外,由于相关数据的可用性有限,要定义异基因CAR-T细胞的有效剂量并不像自体细胞那么容易。从不同供体获得的这些产品的个体差异也应被视为异体CAR-T细胞的缺陷。
Poorebrahim M, Sadeghi S, Fakhr E,et,al. Production of CAR T-cells by GMP-grade lentiviral vectors:latest advances and future prospects.Crit Rev Clin Lab Sci. 2019 Sep;56(6):393-419.