在人体中,基因表达是动态的,但也受到严格地调控。细胞使用多种机制来控制单个基因在何时、何处启动和关闭。而当这些机制失灵时,由此引发的异常基因表达往往会导致癌症。
在DNA和DNA相关蛋白上添加各种化学标记是控制基因表达的一种方法。近期,发表在Nature杂志上的两篇论文[1][2]显示,控制这种标记添加的名为NSD3的酶在一些人类癌症中发生了突变,变得极度活跃。这些发现为治疗这些癌症提供了潜在的新途径。
在细胞中,DNA缠绕在被称为组蛋白的蛋白质簇周围,形成染色质基本结构单位――核小体,每个核小体由146bp的DNA缠绕组蛋白八聚体1.75圈形成。DNA和组蛋白可以进行广泛的修饰,各种化学基团被共价添加到DNA碱基和组蛋白的尾部,或从DNA碱基和组蛋白的尾部移除。这些变化被称为表观遗传修饰:它们并不改变基因序列,而是构成一种信号,告知细胞哪些基因应该被打开和关闭。
添加或移除表观遗传修饰的酶通常在癌症中过表达或突变,这表明它们在癌症发生和维持中扮演着角色。不过,调控表观遗传修饰的酶的失调导致癌症发生以及维持癌症发展背后的生化机制尚不清楚。最新发表在Nature杂志上的两项成果部分解答了这一问题。研究揭示了NSD3发挥促癌功能背后的机制。斯坦福大学的生物学教授Or Gozani博士是这两篇论文的共同通讯作者。

来源:Nature
NSD3是核受体结合SET域蛋白(nuclear receptor-binding SET domain protein, NSD)家族组蛋白甲基转移酶中的一员,该家族还包括NSD1、NSD2。组蛋白赖氨酸甲基转移酶通过催化转移甲基基团到组蛋白H3和H4末端特定的赖氨酸侧链上,形成组蛋白甲基化标记,从而影响基因转录、DNA复制和DNA修复等过程,对维持染色质稳定和基因表达调控具有重要作用。NSD3是一种组蛋白H3第36位赖氨酸(H3K36)甲基转移酶,催化H3K36二甲基化,即增加两个甲基基团。在2021年2月3日发表在Nature上的一篇论文中,Gozani教授及其合作者利用小鼠模型和人类肿瘤样本,将NSD3基因定义为肺鳞状细胞癌的致癌基因,也就说,NSD3的突变导致了失控的细胞增殖。
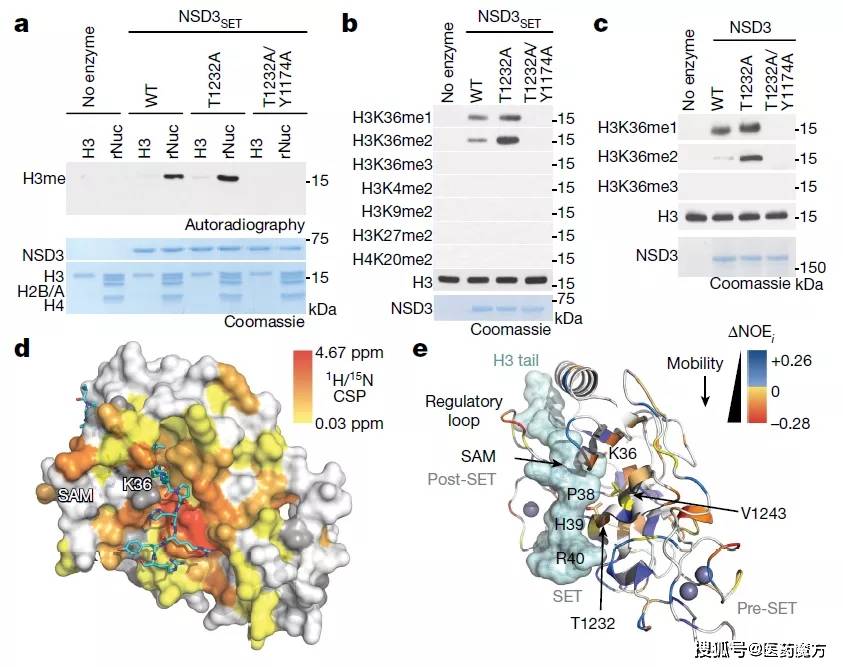
NSD3增加H3K36me2催化的分子基础(来源:Nature)
研究发现,在人类肿瘤中,NSD3变得失调,要么拷贝数增加,要么发生错义突变使其催化活性增加(错义突变指编码某种氨基酸的密码子经碱基替换以后,变成编码另一种氨基酸的密码子,从而使多肽链的氨基酸种类和序列发生改变)。此外,研究显示,NSD3过度活跃的肿瘤中,H3K36二甲基化水平升高。
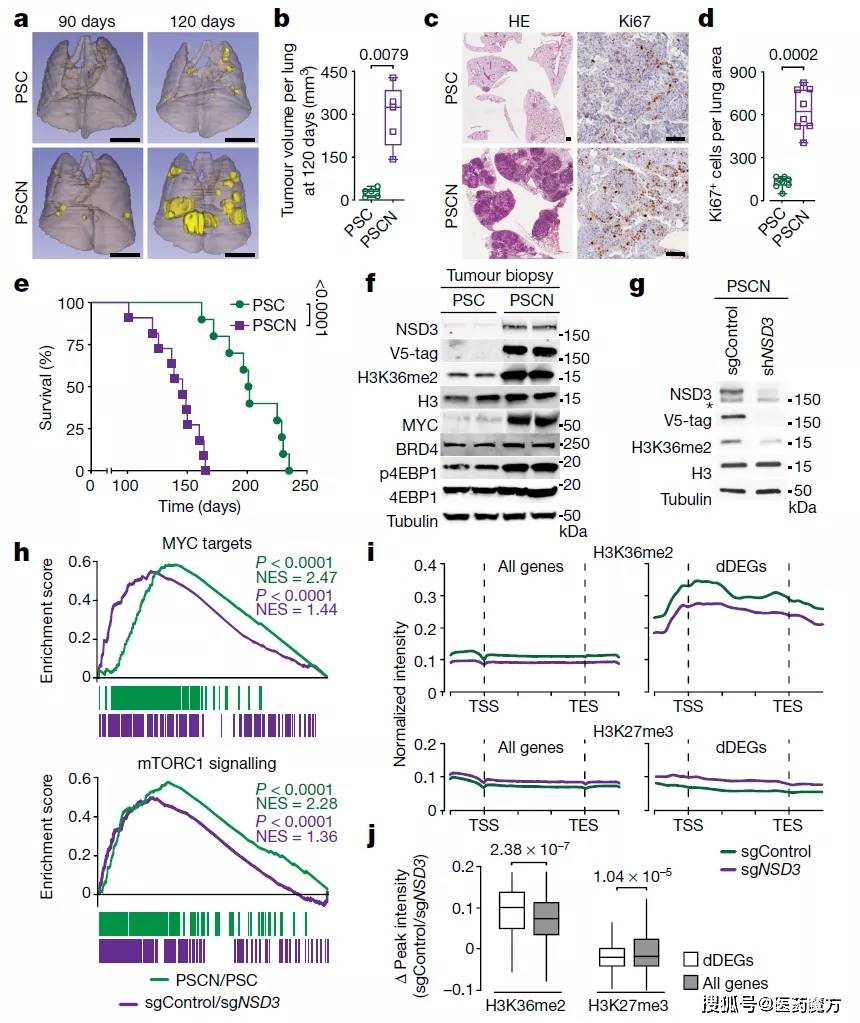
突变NSD3介导的H3K36me2合成促进致癌程序和肺鳞状细胞癌肿瘤发生(来源:Nature)
利用基因组分析技术,科学家们发现,致癌NSD3会催化促癌基因蛋白编码区域的H3K36二甲基化,导致这些基因表达率更高。一组重要的实验表明, NSD3拷贝数的增加(比错义突变更常见)导致肺癌细胞对NSD3的催化功能“上瘾”, 因为NSD3的丢失会通过降低H3K36二甲基化和降低癌症驱动基因的表达来阻碍细胞的生长。这些结果表明,在这些肿瘤中,NSD3介导的催化作用是一个可能的药物靶点。
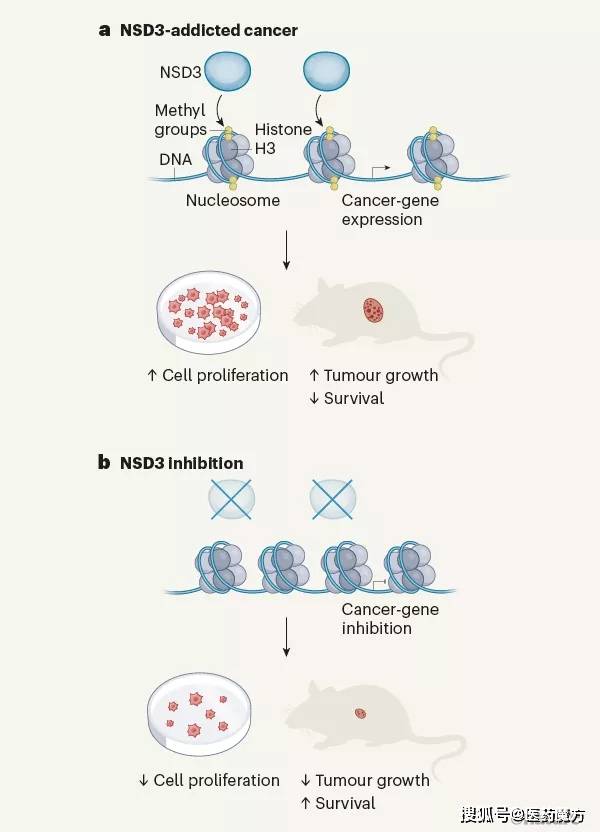
人类癌症中组蛋白甲基化失调 | NSD3是一种组蛋白甲基转移酶,催化组蛋白H3发生二甲基化。Gang Yuan等今年2月发表在Nature上的一篇论文揭示,NSD3在人类肺鳞状细胞癌中失调[1];Wanqiu Li等去年12月发表在Nature上的一篇论文揭示了相关的结构机制[2]。a)在NSD3成瘾癌症中,NSD3的酶活性通过增加NSD3基因的拷贝数,或通过导致酶活性过度活跃的突变而增加。这反过来又会导致组蛋白H3二甲基化和癌症相关基因的表达增加。在细胞培养中,最终会导致细胞增殖增加,而在小鼠中,最终会导致肿瘤生长增加和动物存活率下降。b)抑制NSD3可能为这些癌症提供靶向治疗。NSD3的缺失可导致组蛋白H3二甲基化减少、癌症基因表达下降、培养物中细胞生长减少以及小鼠肿瘤减少。(来源Nature)
在2020年12月发表的一篇相关论文中,Gozani教授及其合作者利用冷冻电子显微镜技术揭示了正常NSD3和致癌突变型NSD3结合到核小体上不同的结构特征。该研究的一个主要贡献是,解释了长期观察到的NSD蛋白催化核小体中H3K36(而不是游离组蛋白)甲基化的偏好。研究发现,在没有底物的情况下,NSD3的底物结合位点会被该蛋白自身的一个环所阻断。与核小体表面以及部分解开的DNA的几次相互作用会使这个环发生移位,为组蛋白H3尾巴进入NAD3的催化位点腾出空间。
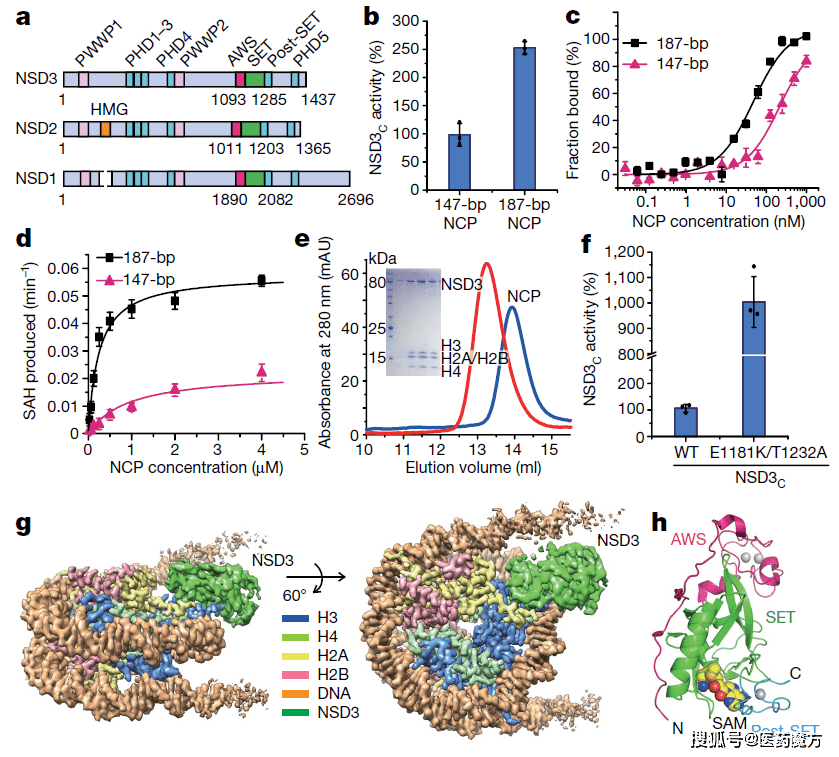
NSD3的生化分析及NSD3C(E1181K/T1232A)-NCP复合物的整体结构(来源:Nature)
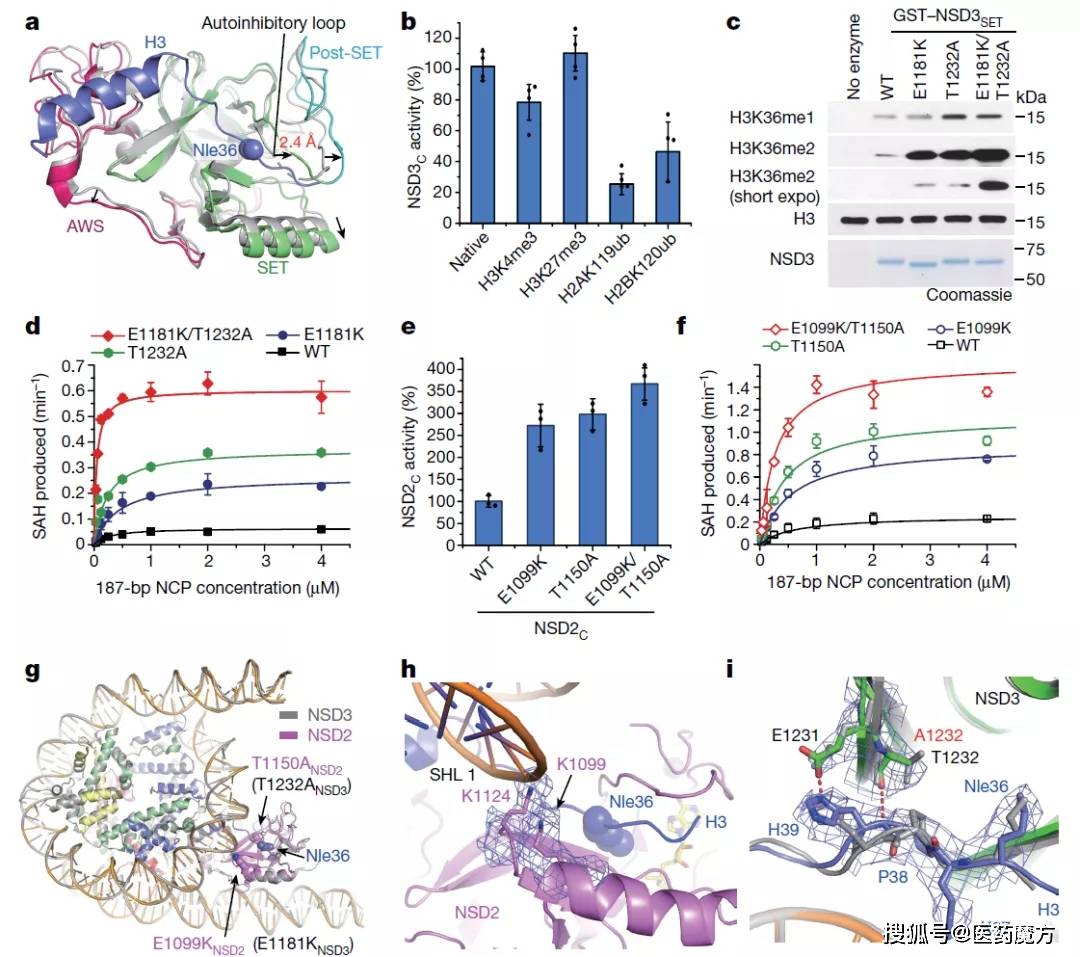
NSD2和NSD3及其癌症相关突变的结构和生化分析(来源:Nature)
结构分析还揭示了NSD3致癌突变如何导致与组蛋白形成新的氢键,解释了突变体为何具有增强的催化活性。这些原子分辨率的NSD3结构,以及新工作中报告的酶学研究,将为鉴定小分子NSD3抑制剂提供基础。
值得注意的是,去年发表在Nature Chemical Biology 上的一篇研究描述了共价抑制NSD1甲基转移酶功能的小分子,这是对“选择性靶向NSD是可行的”这一观点的概念验证[3]。
Nature就这些新发现发表的观点文章认为,抑制NSD3可能成为新的癌症靶向治疗策略。同时,该文章还强调,大量证据指出,H3K36甲基化是癌症表观遗传失调的一个重点,这两篇新论文为支持这一观点提供了进一步证据。
参考资料:
[1] Gang Yuan et al. Elevated NSD3 histonemethylation activity drives squamous cell lung cancer. Nature(2021).
[2] Wanqiu Li et al. Molecular basis of nucleosomal H3K36 methylation by NSD methyltransferases. Nature(2020).
[3] Huang Huang et al. Covalent inhibition of NSD1 histone methyltransferase. Nature Chemical Biology(2020).
[4] An epigenetic tipping point in cancer comes under the microscope(来源:Nature)