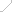һ������ҩ��ӷ��ֵ����յ����У�����Ҫ�����»�ѧNCE�ķ��֡��ٴ�ǰ�о�����ҩ�ٴ����飨Investigational New Drug��IND���걨���ٴ��о���NDA��New Drug Application�����뵽��������Լ����к���˾����Բ�Ʒ����IV���ٴ��о������к���ȵȹ��̣����е�ÿһ�����п��ܻᵼ��ҩ���з�ʧ�ܡ�
ҩ�↑����ҩ��ֺ�ɸѡ��ʼ����ҵ�迪չһϵ�еĺϳɹ��ա��Ƽ����ա������о��ȵ�ǰ�ڹ������һ��迪չһϵ�ж��ﶾ����ҩЧ�ͣ���ҩ������ѧ�о����õ�ҩ�ﰲȫ�Է���ij���֤�����������ٴ����顣
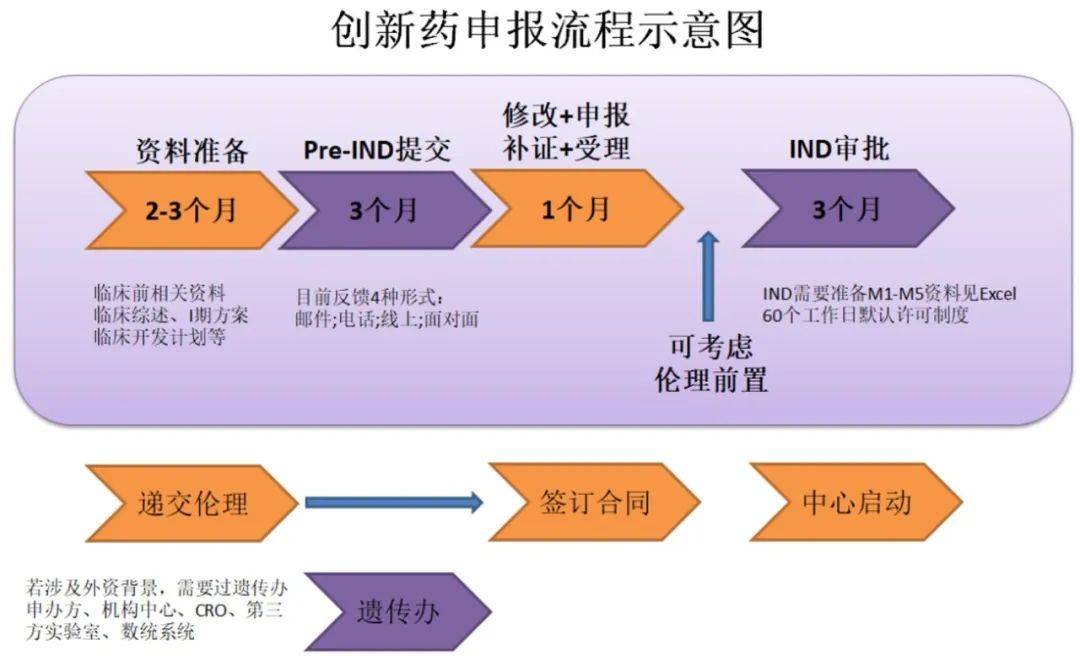
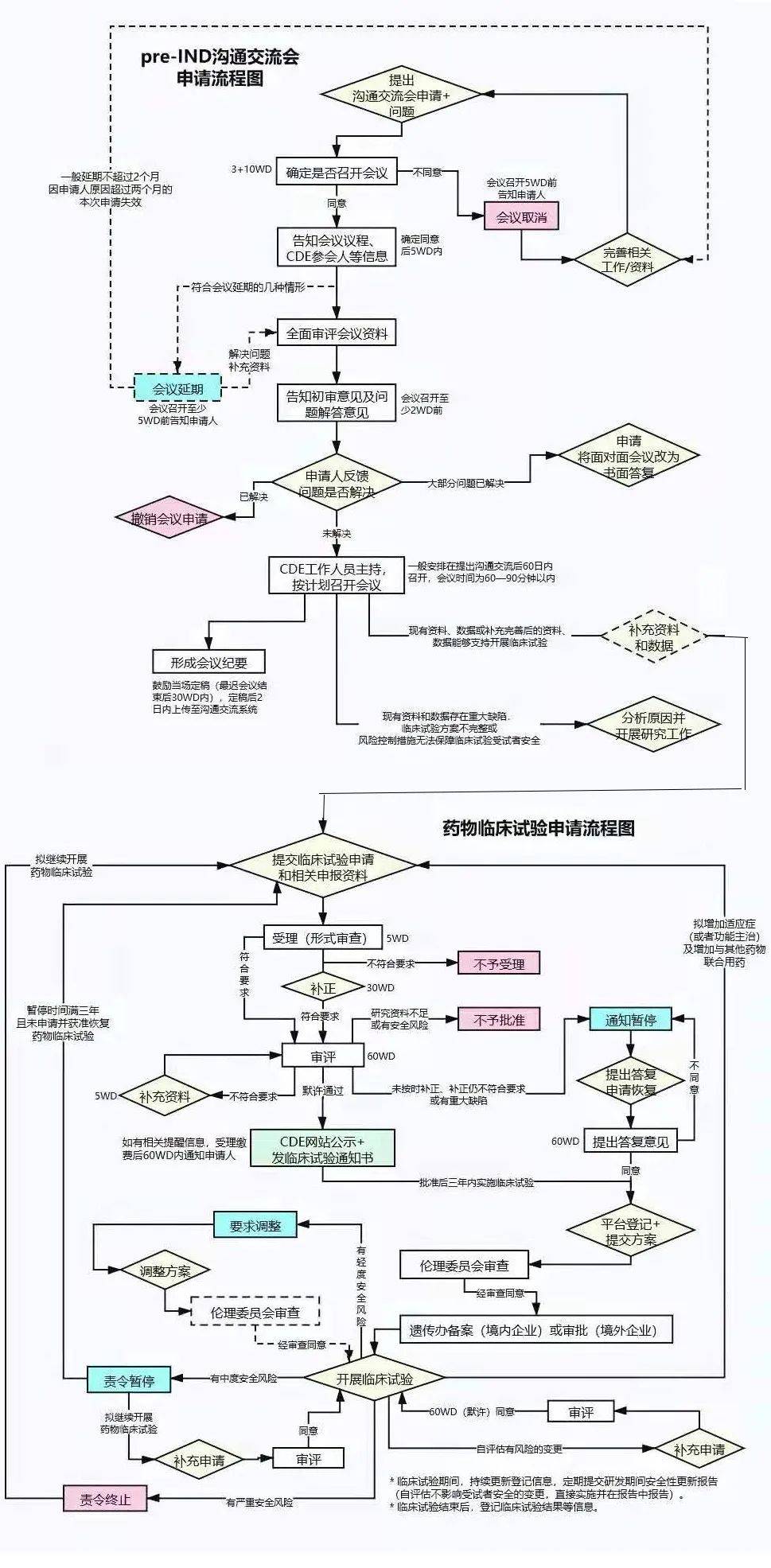
FDA�����˶�����Ч�ĺ���ҵ�Ĺ�ͨ���ƣ������������ҵ����ʽ���飬��Pre-IND���顢�����ٴ���������顢NDA/BLA�ݽ�ǰ����ȵȡ�ͨ�����飬��ҵ���Ի�ȡ����FDA����ҩ�↑������Ľ��顢���FDA��Ҫ��������·���ߴ�·�Ŀ����ԣ�����걨�ɹ��ʡ�����˼�壬���걨IND֮ǰ��ҵ��FDA�Ļ��飬����ΪPre-IND���顣
Ϊ�ˣ���ҵ��Ҫ��һ��Pre-IND package��Ԥ�ȸ�֪FDAҩ��Ļ�����Ϣ���о���״���������о��ƻ��ȵȣ�ͨ������CMC���ٴ�ǰҩ���������ٴ����鷽����١����������ٴ����飨���У����������ϣ�������Ҫ�����ݵ�����ҵ����Pre-IND��������FDA���۵����⡣����ʲô�����������˵������Pre-IND������Ҫ��һ�����
һ��˵������ҵ���ڼƻ���FDA����ǰ60�����ң���FDA����������롣FDA���յ����������һ����14�����������鰲�š�֮����ҵ��FDA�ݽ�Pre-IND���ļ���һ���ڻ���ǰ4�����ҵݽ�����FDAͨ����ظ�������������ij����������ҵ�����˽FDA������ij���������ͨ����ҵֻ��һ����FDA��Pre-IND����Ļ��ᣬ����Ҫ�Բ���С�
��Pre-IND������������������գ�FDA���ṩ��ʽ��Pre-IND�����Ҫ����¼FDA���걨�߶����������������۽�����걨��Ҳ�����ڻ���ṩ�Լ��Ļ����¼��FDA�������Լ���������������⣬����˫�������ϵ���
03 IND�걨�ļ���
����Pre-IND������FDA�����۽�����걨�߽���IND�걨�ļ����ı�д��
IND�걨�ļ�����Ҫ����9���ֵ����ݣ�����ҳ����FDA 1571������Ŀ¼�������Ժ������о��ƻ������о�Ա�ֲ���ٴ��о���������ѧ������������������Ϣ����ҩ���Ͷ�����Ϣ�������������ٴ����飻�������Ϣ����Ҫע����ǣ���IND�걨�ļ����У������ύ���ԭʼ�����о����棬�綾���о�����ȡ�
04 IND����
����������FDA�յ�IND�걨�ļ�����ʼ��ʱ��FDA����30����������������������ڣ�FDA���ܻ���걨�߾�ijЩ���չ���ۡ��������һ��������������������ʼ�ٴ��������ٴ������Լ��ٴ����ơ�
IND�걨�����̱Ƚϸ��ӣ���������IND������ÿһ����������ʧ�ܵĿ��ܡ��б�����IND����ʧ���ʳ�����90%������ҩ���з��걨ʧ�ܵ�ǰ3λ���طֱ���ҩ������ѧ���ٴ���Ч�ԺͶ����о���
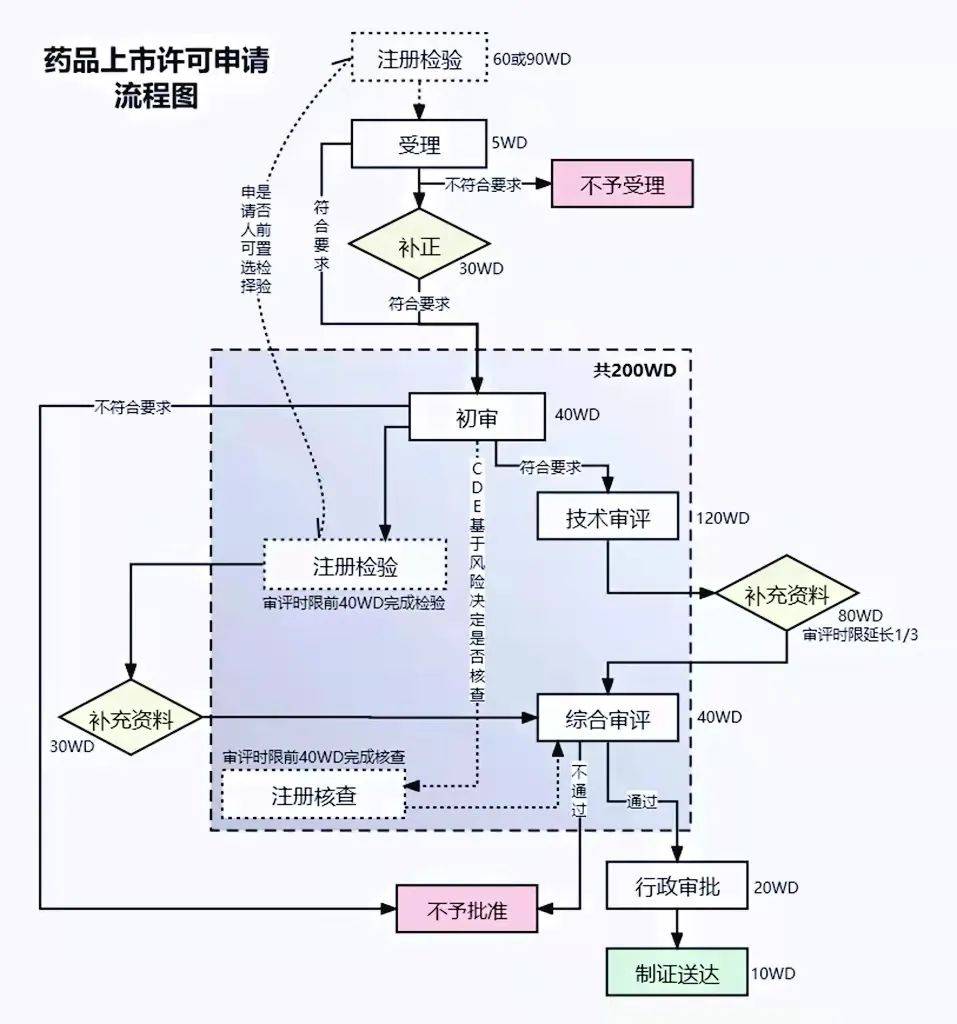
05 ҩƷ���к���������