我们都知道,肿瘤细胞在疯狂增殖过程中遗传信息会发生改变,不断获得些奇奇怪怪的功能特性,但就是不干正事儿。
近日,一篇发表在《自然》期刊上的论文揭示,在肿瘤抑制因子p53的管教下,肿瘤细胞的胡乱行为得到约束,转而向安分守己的方向发展。
美国斯坦福大学的Laura D. Attardi及其同事们发现,p53调控着肺部细胞的再生分化程序。在肺腺癌进展过程中,肿瘤细胞自身表达的p53,能够促使其分化为功能接近于正常肺部细胞、活性较低的肿瘤细胞类型[1]。
 论文首页截图
论文首页截图肺癌是全球癌症死亡的主要原因,最常见类型为肺腺癌。约有一半的肺腺癌患者携带失活突变的TP53(p53的编码基因),导致肿瘤侵袭性增强和预后恶化。然而,p53如何抑制肺腺癌一直不为人所知。
在这项研究中,Laura D. Attardi等人利用遗传学手段,构建p53功能活性不同的肺腺癌小鼠模型,并与p53正常、未发生突变的肺腺癌小鼠模型进行对比。这些小鼠均携带基因Kras(G12D)激活突变。
结果显示,与p53未突变患癌小鼠(KT)、p53失活患癌小鼠(KPT)相比,p53过度活跃的患癌小鼠(KFT)肿瘤负担减少,肿瘤进展受到抑制。同样,病理学分析结果提示,p53活性增加与人类患者肿瘤恶性程度降低有关。
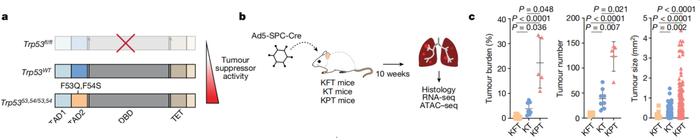 p53活性越强,肺腺癌小鼠肿瘤负担越低
p53活性越强,肺腺癌小鼠肿瘤负担越低这些结果证明了p53在肺腺癌发生和进展中的重要性。另外,小鼠实验中还观察到,p53仅在肿瘤细胞而非间质细胞中表达,说明p53的肿瘤抑制活性由肿瘤细胞自身表达的p53来驱动,不依赖于外部信号或邻近细胞的影响。
p53究竟是如何驾驭肿瘤细胞的呢?
作为一种转录因子,p53能够结合到DNA上特定的序列区域,通过调节基因的转录来影响细胞功能和生理过程。这意味着,p53手下管控着众多基因的表达。
从这个思路入手,研究者们利用单细胞RNA测序等技术,对小鼠肺部细胞的基因表达模式进行分析。结果发现,上述小鼠模型的肿瘤细胞具有明显不同的转录谱,有超过5000个与p53相关的差异表达基因。与p53功能缺失的细胞相比,p53功能正常或过度活跃的肿瘤细胞中,参与调节细胞发育和分化途径的基因表达水平增加,肿瘤细胞维持着类似正常肺细胞的特征。
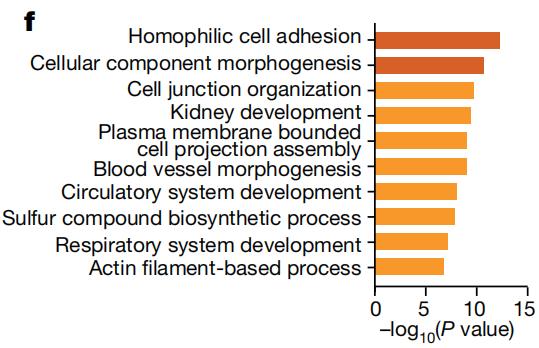 肿瘤细胞内p53活性增强时信号通路的富集程度
肿瘤细胞内p53活性增强时信号通路的富集程度随后,研究者们发现,p53是在帮助肺腺癌细胞分化为更加稳定的肿瘤细胞类型。
肺部中,I型肺泡细胞(AT1)和II型肺泡细胞(AT2)共同构成肺泡结构。薄而扁平的AT1细胞功能高度特化、不再增殖,担负着气体交换的重任;立方形的AT2细胞则主要负责分泌活性物质,且具有干细胞特性,能够自我更新并分化为AT1细胞。肺部损伤修复时,由AT2细胞增殖分化来补充AT1细胞。与此同时,还有处于介于中间状态的过渡细胞。
值得注意的是,肺腺癌起源于AT2细胞,并且随着肺腺癌进展到不同阶段,肿瘤中存在AT2样细胞、AT1样细胞以及过渡细胞。
在这里,利用肺腺癌小鼠实验,研究者们发现,p53功能活性越强的小鼠肿瘤内AT1样细胞水平更高。与AT2样肿瘤细胞相比,表达p53的AT1样肿瘤细胞中,与肿瘤进展相关的基因表达降低,并在增殖过程中遗传信息相对稳定传递、保持类似正常AT1细胞的功能和特性。
如果诱导肿瘤细胞表达功能正常或过度活跃的p53,可以通过细胞形状和标志物表达情况观察到,肿瘤细胞向AT1样细胞转变。
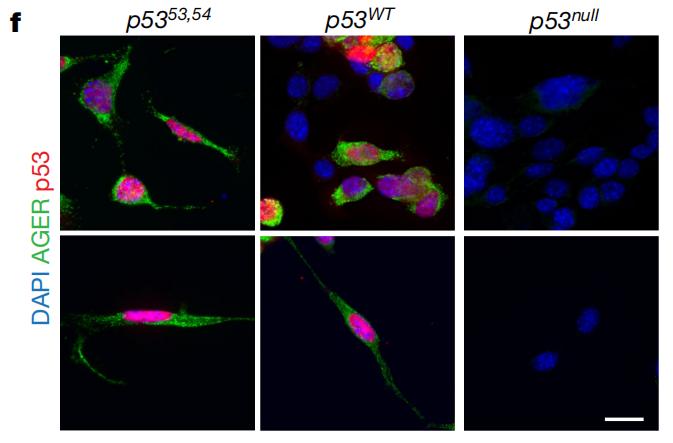 从形状来看,表达p53时,肿瘤细胞向AT1样细胞转变
从形状来看,表达p53时,肿瘤细胞向AT1样细胞转变对多组肺腺癌患者队列的数据进行分析可以得到相似的结果。与TP53基因发生功能性缺失突变的患者相比,具有完整TP53基因的患者肿瘤中AT1样细胞水平更高。
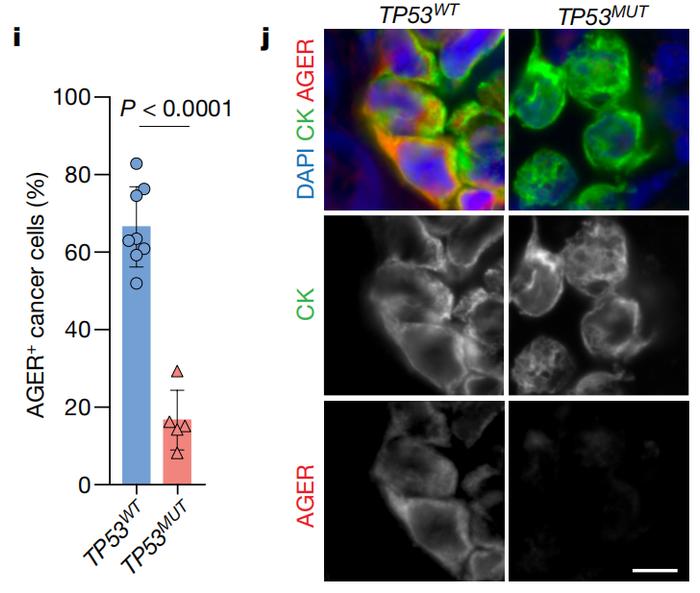 从AT1细胞标志物来看(AGER),携带TP53突变的肺腺癌患者肿瘤中AT1样细胞更少
从AT1细胞标志物来看(AGER),携带TP53突变的肺腺癌患者肿瘤中AT1样细胞更少鉴于p53是个转录因子,研究者们还通过染色质免疫共沉淀实验找出与p53发生互作的DNA序列。结果发现,p53结合的DNA序列区域包含大量AT1细胞相关基因,而非AT2细胞基因。而且,这些受p53关照的AT1细胞相关基因更容易被细胞转录和表达。
也就是说,肿瘤细胞中,p53直接参与AT1细胞特性的激活,从而促进肺腺癌细胞向AT1样细胞方向分化。
除了抑制肺腺癌进展,研究者们发现,p53以同样的方式帮助肺部损伤修复,即通过指导AT2细胞向AT1细胞分化、补充AT1的数量来恢复肺部正常功能。
具体而言,在AT2细胞→过渡细胞→AT1细胞的分化过程中,每一步都得有p53的监督。
一方面,p53会抑制AT2样肿瘤细胞或AT2细胞的自我更新,促进其启动分化为过渡细胞。
另一方面,过渡细胞就好像处于“人生岔路口”,面临多种分化方向。p53则会促进过渡细胞继续向AT1样肿瘤细胞或AT1细胞分化,限制其恶性分化,从而抑制肿瘤进展或阻止组织进一步损伤。
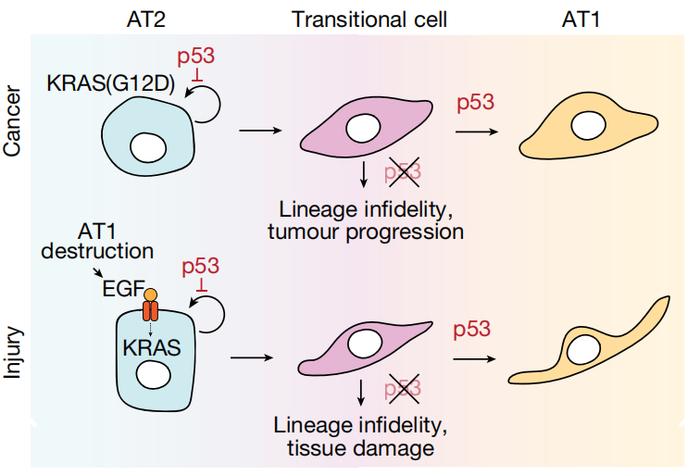 机制图
机制图总体来说,这项研究从一个十分新颖的角度揭示了p53抑制肺腺癌的机制。
有趣的是,同期发表在《自然》期刊上的另一篇文章发现,致癌驱动因子KRAS的操作正好与肿瘤抑制因子p53反着来。
刚才不是说,p53能够诱导AT2细胞分化为AT1细胞吗,美国斯坦福大学的Tushar J. Desai等人的这项研究则是揭示,KRAS(G12D)突变能够对细胞进行重编程,诱导AT1细胞回炉重造――去分化成为AT2细胞[2]。
不仅如此,Tushar J. Desai等人也将AT1细胞鉴定为肺腺癌的起源细胞。他们发现,在肺癌发生和进展过程中,异常AT2细胞往往具有更高的增殖活性,能够迅速扩张、具有侵袭性,异常AT1细胞增殖相对缓慢。由AT1细胞恶性转化成的肿瘤在早期较温和,但随着疾病进展、KRAS(G12D)突变的重编程作用,肿瘤内AT2样细胞增多。
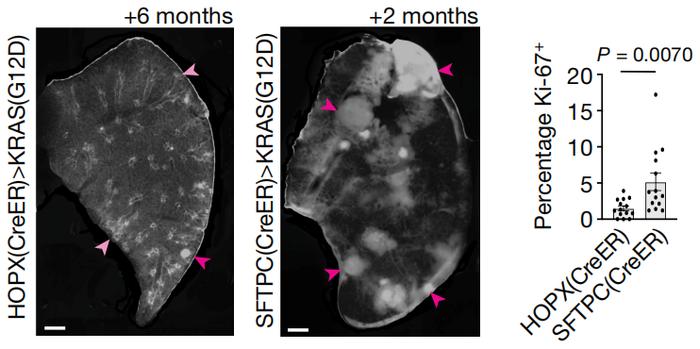 起源于AT1细胞的肺腺癌,在发展6个月时(左)、由KRAS(G12D)诱导2个月后(右)的组织学、分子特征
起源于AT1细胞的肺腺癌,在发展6个月时(左)、由KRAS(G12D)诱导2个月后(右)的组织学、分子特征如此看来,天平倒向AT1细胞还是AT2细胞,是属于肿瘤抑制因子和致癌驱动因子之间的博弈啊。
参考文献:
[1]https://www.nature.com/articles/s41586-023-06253-8#
[2]Juul, N.H., Yoon, JK., Martinez, M.C. et al. KRAS(G12D)drives lepidic adenocarcinoma through stem-cell reprogramming. Nature (2023). https://doi.org/10.1038/s41586-023-06324-w
发布于:北京