�����ҿ���������Ȼ����־�����Ͳ�Ӧ�÷����ȸ����ƪ���ģ���Ϊ��Υ����FAIR��Findable�ɷ��֡�Accessible�ɷ��ʡ�Interoperable�ɻ�������Reusable�����ã�������ԭ�����ȸ������������������ģ�͵����ݣ�����������ģ�ͽ��������Ψһ������������ģ������ʶ������ȶ����壬���������Ը���ģ�͡���������Ϊ����ȸ������Ĺ�˾�����ѧ���̹�Ȼ��Ҫ����Ҳ��������ͬ�����Ͻ��������۴��ĸ���������һ��������֤�Ĺ��������ܱ���Ϊ��ѧ����
����Shyue Ping Ong��UCSD���ڣ�Materials Project�����ˣ�
��ͷ������AI+���Ͽ�ѧ��
2023��11�µף�Google���µ�DeepMind��Nature��־�������ذ����ģ��������ǿ��������ڲ��Ͽ�ѧ���˹�����ǿ��ѧϰģ��Graph Networks for Materials Exploration ��GNoME������ͨ����ģ�ͺ�ͨ����һ��ԭ�����㣬Ѱ�ҵ���38���������ѧ�ȶ��ľ�����ϣ��൱�ڡ�Ϊ����������800����������ۡ�������ӿ��˷����²��ϵ��о��ٶȣ�ͼ1����[1]

ͼ1. Google���µ�DeepMind��Nature��־������GNoME���ݼ���ģ�͡�
2023��12�£�����Google��GNoMEģ�ͷ���������������˲��Ͽ�ѧ������˹���������ģ��MatterGen���ɸ�������Ҫ�IJ������ʰ���Ԥ���²��Ͻṹ�����ܲ����罻ý����Ϊ�ԼҴ�ģ��վ̨�����۵����������з���MatterGenģ�Ϳ��Դ�������²��ϵİ����з�Ч�ʡ���ͼ2����[2]

ͼ2. ���ܲ������Լ��˹����ܲ�������ģ��
2024��1�£�����������Դ������������̫ƽ�����ʵ���ң�PNNL�������������˹����ܺ����ܼ��㣬��3200������������ɸѡ����һ��ȫ��̬����ʲ��ϣ�����˴�Ԥ�ʵ��ıջ����ü�����������һ������ӵ�ز����з���ͼ3����[3]

ͼ3. ���Ŀ�ѧ�Ҵ�3200������������ɸѡ��ȫ��̬����ʲ��ϣ���ʵ����֤��
���Ͽ�ѧ������һ����Ҫ��ѧ��Ҳ�ǽ�����ҵ���ٷ�չ��֧��ѧ�ơ���ʯ��ʱ������ͭʱ�����ٵ�����ʱ�������������ĸ����ݻ��ζ��Ͳ��Ͻ�����ء��մ�Ϊ���������ķ��������˺������ͣ������ٳ��˹�ѧ�����ķ�����Ϊϸ������ѧ������ѧ�Ľ��������˷��ʡ�����˵������������չʷ����һ�����Ͽ�ѧ���ݻ�ʷ��
���ڣ��˹����ܼ����Ľ�����ν��һ��ǧ��������𡣽��˹����ܷ�����������ѳ�Ϊһ����Ҫ�Ľ���ѧ�Ʒ�����Google������Meta���ֽ�����Ҳ�ڽ��ڲ��������Ƶ��з�����Meta AI��������У��������������ҵ�����Ĵ��������ݼ�Open Catalyst Project���л���������������ݼ�OpenDAC��һʱ�䣬�Ƽ���ͷƾ���Լҵļ����������Ͽ�ѧ���������������ӿ�������Ͽ�ѧ��Ϊ�����ǵ���������
//
GNoME���Ͽ�ѧ���ݼ���ϸ���
�˹���������α������з��ģ��Ƽ���ͷ�ǿ�����ͬ�ļ���·�ߣ���1��ͨ�����ۼ����ȡ���Ͽ�ѧ���ݣ���2��ͨ����ͨ���������������������ݣ���3���ٽ�����ι���˹�����ģ�ͣ���4������ģ������δ֪���ϵ����ܡ���Ҳ��ζ������һ����֮��Ч�ļ���������ǰ��������
�˹������Ƿ��δ�����Ͽ�ѧ���з���ʽ�����ǿ϶��ġ����ݡ��㷨������Ҳ����Ϊ�ٳ��ⳡ���ĺ������ء�������ǵص����ź������У���������Google���������ݼ�Ϊ����㣬������ϸ���ݺ���һ̽������
1. ������ҽҩ��ҵ֮���Ͽ�ѧ���˹����ܴ�ٽ������һ����ڡ�����ǰAI�����������ҩ����������Schrödinger��˾��Atomwise��˾���ڶ���ҵ��������ģ������ҩ��ҵ�������»��ᣬ��ԭ�ӳ߶�ɸѡĿ��ҩ����ӳ�Ϊ�˸���ҩ���з������е���Ҫһ����
Ȼ��ҩ���з����ڳ����з��ɱ��ߣ����������ϸ�������в���AI��ҩ��˾תս���Ͽ�ѧ������Schrödinger��˾�����˲��Ͽ�ѧ���š������ϣ�����������ҽҩ�������ʿ�ѧ��AI���ܱ��������һ�µģ�ͨ���˹����ܷ������ҵ�ԭ�Ӽ�����õ��������ģ������
�Ƽ���ͷ����ʶ�������Ͽ�ѧ����ҩ������ͬ�ĵײ��������¾߱���ֻǷ�����ݡ����������˹�������ɵ����Ƽ������ݼ��Ĵ�С�������ߵ�ֱ�Ӿ������˹����ܵ�Ԥ�Ȿ�졣���ڣ������ڲ��ϻ��̺����ɲ��Ͽ�ѧ���ݿ�ķ�չ���������Ѿ߱����ʵ�������Դ���˹����������ǰ�������Ѿ��̵���ˡ�
2. ���ݼ����˹����ܴ��õĵػ����˹�������������ݵ������ȼ��ߣ����ݼ��ĸ��ǶȺ�����ֱ�Ӿ������˹�����ģ�͵ĸ߶ȡ����ݼ��ĸ��ǶȾ�����ģ�͵ķ������죬���ݼ���һ���ԺͿɱȽ��Ծ�����ģ�͵�Ԥ�⾫�ȡ����˹��������ݡ��㷨����������Ҫ���У���������������ݵĻ��ڡ����磺GPT 3.5��Llama 2�ȴ�����ģ�ͣ���ʹ��Դģ��Դ���룬Ҳ��ѡ���������ݼ���û����������ݼ���Ϊ֧�ţ���ҵ�еľ������ֺ���ѵ���������AIģ�͡�
�㷨�Ѿ���ʧȥ�˼����������ã�ƾ���㷨������ҵһ֦����Ŀ�����������
3. ���ۼ���Ϊ�������Ͽ�ѧ���ݿ������˺������͡��ܶȷ������۾�����ʮ��ķ�չ���ѻ����˳���ļ��������������ڶ�ʱ�����������߶ȱ��������ݼ����ܶȷ�������ͨ�������ϵ�еĵ����˶����̣����Ը�Ч��������������ʣ��Ӷ�������������ԭ�ӿռ�ֲ��뻯�������Ե���ϵ��ͨ��ͬʱ���гɰ���ǧ��������ҵ�����DZ�������������������ݼ���Ŀǰ���Ͽ�ѧ����ʹ����㷺�����ݼ�����Materials Project[4]��OQMD[5]�����ǻ����ܶȷ������۸�ͨ�������õġ�GNoME���ݼ���ζ��Google�Ѿ������˲��Ͽ�ѧ����������������
����Ŀǰ�IJ��Ͽ�ѧ�з���������ƾʵ�����ݻ��ۣ��������ڶ��������Ƶ����ݸ��ǶȺ�һ���ԡ�
4. Google�����İ�����GNoMEģ�ʹ�������ݼ������֡����ݼ����ǶȺ;��ȷdz��ߡ�GNoME���ݼ���Materials Project������ã���������Materials Projectһ�µļ�����ͼ������̣���˿��Ժ�Materials Project[4]�ϲ�ʹ�á�Google����ͨ����ͨ��������ܶȷ�������������220���������ϵļ������ݣ������ͬʱͨ������ѧϰ����Ԥ������ѧ�ȶ����²��ϣ������ҵ���38�����ȶ�����������������ǶԲ��Ͽ�ѧ����ľ��ƶ���
5. ��ȻGoogle���յ�GNoME���ݼ��ܴ���220���������ϣ����������Ĺ�������Ϣ��������С���ֵ����ݣ���38������������Ľṹ������ѧ�ȶ��Լ�ģ�ʹ��롣Google����δ����ģ�Ͳ�������˵��������Կ��伴�õķ�ʽ����ģ�͵�������GoogleҲû�з������������ݣ�������ͨ�������ݼ���չ��Ч��ģ��ѵ������ˣ�Google������GNoMEģ�͵Ķ�ռ����
��δ����AI��ģ�ͽ��������У������ǻ��Ǻӣ�Google����Դ�������ݣ���֤������ҵ�в��ɱ���Խ�������λ����ʹGoogle������38���������Ľṹ������ѧ�ȶ��ԣ�����google��û�й����ܶ�ؼ���Ϣ�����绯������γ��ܣ�formation energy������ƾ�ѹ�����38����������ݣ���������Ҳ��ѵ�������Ч��ģ�͡�
�������ɻ��������ʱ�����Ļ��ڣ�����Ŀǰ��ҵ�����ӭ�Ѷ��ϣ�ͨ�����ַ�ʽ�����������ݵĻ�������֯���о��߷dz����ޡ���������ڴ���ˡ�˳�糵������Ҷ����ݹ��������ڴ���ȴ��ͼ�ܿ��������������⡱��
Ϊ�˽��������⣬��ҵ��һ�����еĹ��������ݻ㽻���Ѹ��ֹµ����ݡ���ϡ���һ���γ�һ������һͳ�������ݼ����������ʣ�����һ�ּ�ϣ���������������ݵķ�ʽ������ǰ�ͱ��Ƴ磬����Ŀǰ��δ�����ɹ����������磬�Ƽ����IJ���ר��������Ƶ����ݻ㽻���ơ�
���ɿƼ���ͷ�����ѵģ����Ǻ��������ӭ�Ѷ��ϣ��Լ��������ݡ����Ǵ����Ҳû����Ը�������ع�����Щ��������ݼ�����Ҳ�Ǻ�������ģ���Ϊ��Щ����Ҳ���̺��ž����ҵ��ֵ�������ǶȽ�����Դ�����ݻ㽻�ij������Ч��δ�ض�������ġ�
6. �����ϵ���ռ������ֻ������һС���֡�����������ϸ������������38���ֻ�����Ľṹ��Ϣ����������30345�ֲ��ϵ�Ԫ����ϣ�������Zr-Ti-Se������Ni-Te�������Դ�Materials Project���ҵ���ռ��7.8%������ζ����������֪�Ļ�ѧ�ռ��У�Google�ҵ���30345������ѧ�ȶ��IJ��ϡ����֣�92.2%�����ȶ���������������δ�����Ԫ����ϣ����磺��Rh-Ac������Zn-Cs����������ζ����δ֪�Ļ�ѧ�ռ��У����кܶ�δ�����ֵ��ȶ������������֪�IJ���Ҳ��ֻ�DZ�ɽһ�ǡ����Ƕ�������δ����Ļ�ѧ�ռ䣬���дֻ����ﺬ�еͷ��Ԫ�أ�������ϵ�Ӧ�ü�ֵҲ�Ǵ��ɵġ���ͼ4��
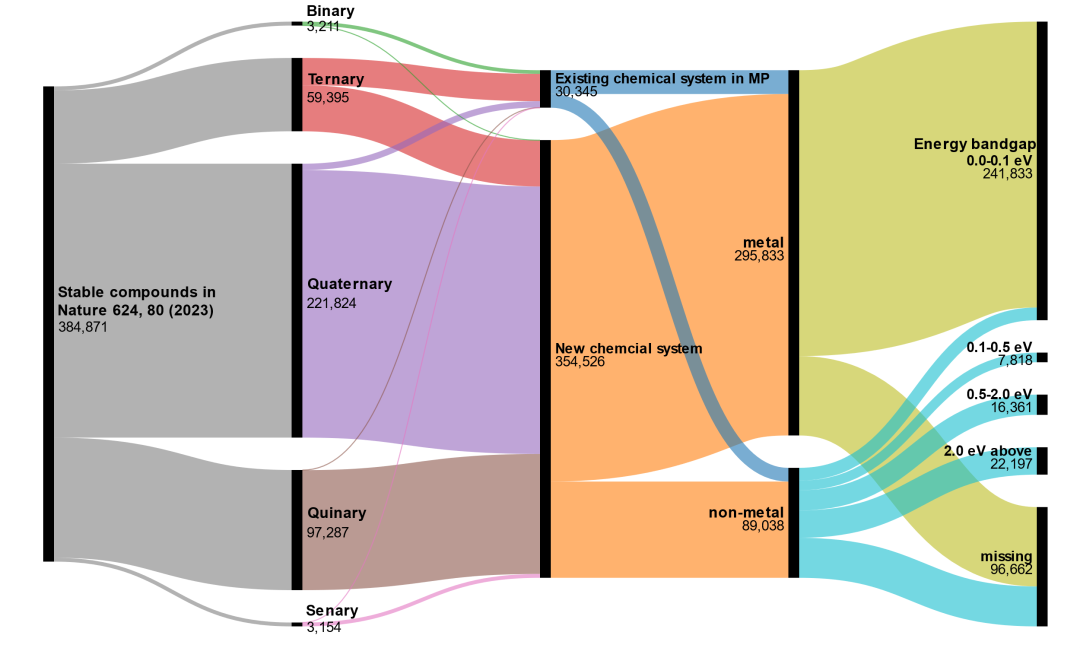
ͼ4. Google��GNoME���ݼ���ϸ������GNoME���ݼ������ҵ���384781������ѧ�ȶ��������ϡ����Կ�����Щ�������У���Ԫ����Ԫ����Ԫ���������������ֻ���������������������Ԫ����ϣ��Ҵ��ǽ��������
7. GNoMEģ���ڸ������Ļ�ѧ�ռ��в����������ݼ������˸������Ľṹ�ռ�ͻ�ѧ�ռ䣬�����һ�����ӡ����ס������ݼ�����Կ�����AIģ�ͷdz����档AI��ģ���̵ı�����һ�֡���ƽ����������ҵ�������������AI�����������ݼ���ڲ壬���������ơ�
����ں���һ��AIģ�͵ĺ��뻵ʱ��ͨ����ָ����Ԥ�⾫�ȣ�����������̸ģ�͵ķ������졣��Ȼ��������ĺû�Ҳ���������궨����߷������죬��Ҫ�����㷺���������ݼ���
�����ҵ��ͨ����Materials Project����Ϊ����������AIģ�ͣ���CHGNET[6]��m3gnet[7]����GNoMEģ��ӵ�и�����һ���������ݼ���������Ȼ���е������ķ������졣
8. GNoME���ݼ��ǡ�����ƫ�ơ��ģ���������ռ��60%���ϡ��Ͻ���ϴ��ںܶ�δ֪�ȶ��ṹ�Ǻ������Ľ������Ϊ����ԭ��֮��������γɽ�����������������ϵ���������Ǻܳ���������Ȼ����Щ����Ԫ������ʵ�����д�����γ�ԭ������ֲ��ĺϽ��࣬����GNoME���ݼ��еĽ����仯���intermetallic������˴���������ϳɡ���ͼ4&ͼ5��
ʵ������У�����Ҽ�������Ԫ�ػ�ϣ�����ʶ������γ�����ѧ�ȶ��ĺϽ𣬵�������Ƿ����²���������㣬���ºϽ��о���С�����ÿ�춼�ڷ��ֳ�ǧ������²��ϡ�
�������˹�����ģ��ѵ������Щ���ݻ������ش�����ġ�
��a�� GNoME
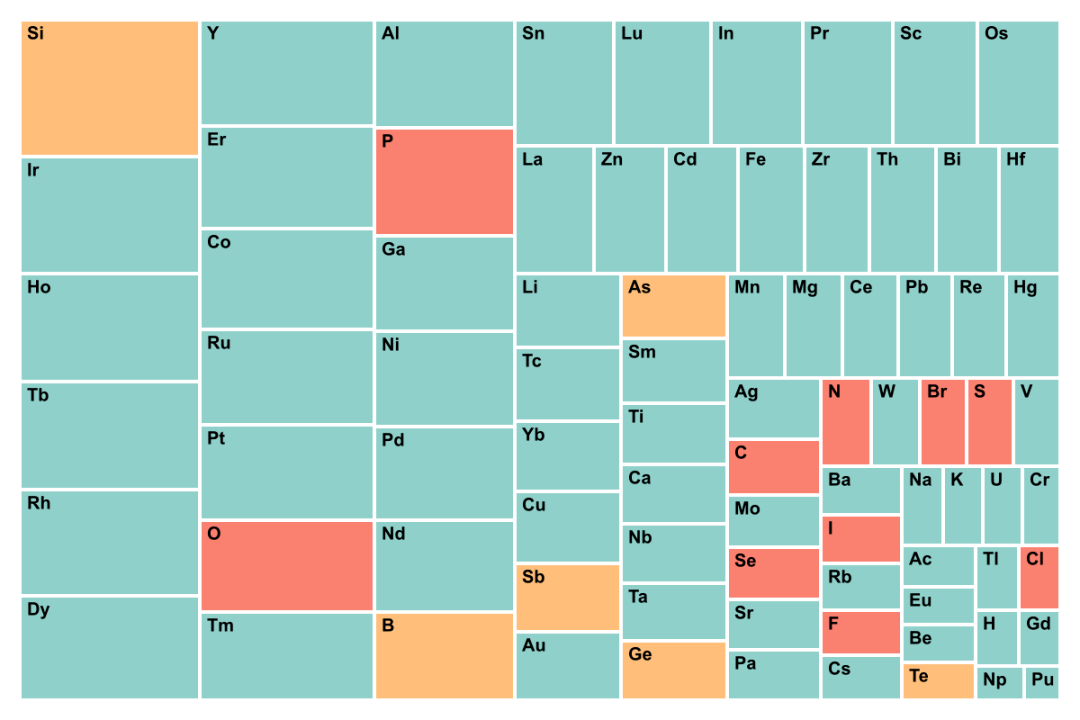
��b�� Materials Project
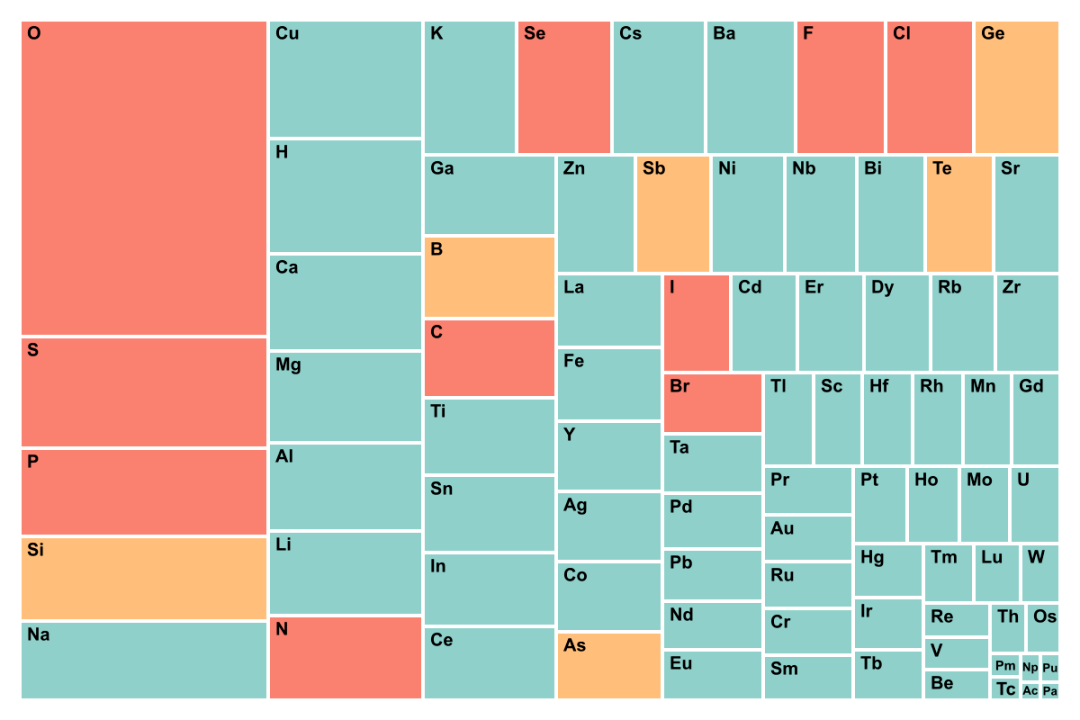
ͼ5. GNoME���ݼ���Materials Project��Ԫ�س��ָ��ʵ�ͳ�ơ�GNoME������Ҫ̽���ͷ��Ԫ�أ��������������Ļ�ѧ�ռ䡣Materials Project̽���IJ�����ϵ�ǽϳ�����ѧ�ռ䡣
9. GNoME���ݼ���Ԫ�س��ִ���ͳ�ƺ�Materials Project��ͳ�ƽ���dz���ͬ��GNoME���ݼ������ӻ�����������٣�������Ԫ�أ��ر��ǵͷ��Ԫ�س��ֵĸ��ʽϴ���Ho��Tb��Rh��Er�ȳ��ֵĴ����ܶ࣬������Ԫ�أ���O��P��S���ֵĸ��ʽ�С�������˵���ˣ�GNoME�IJ����ռ�����ƫ�ĵġ���ͼ5��
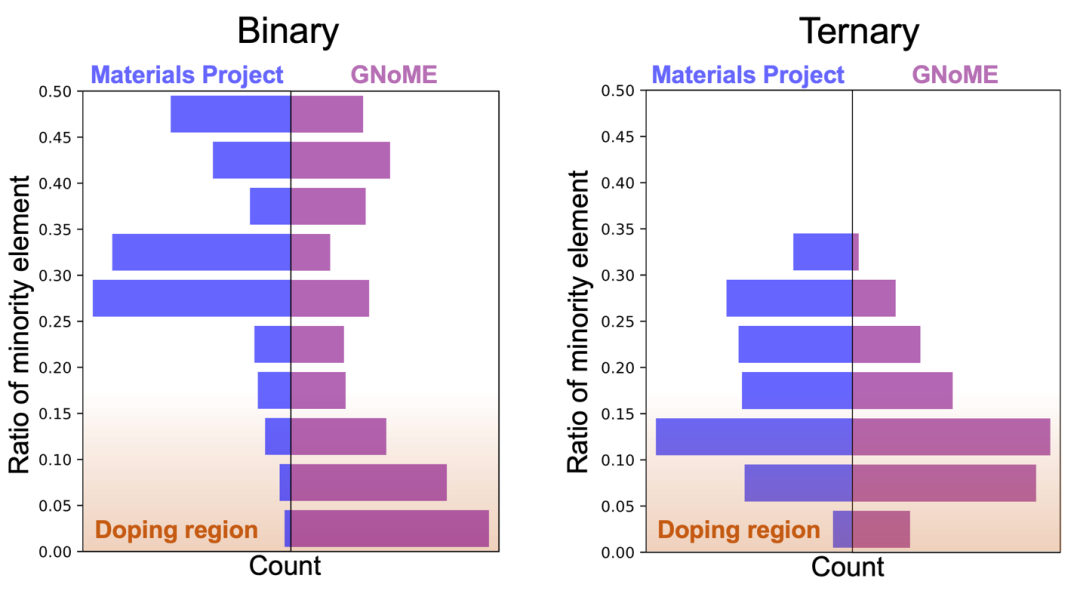
ͼ6. GNoME���ݼ��У�ռ����͵�Ԫ�ص�ԭ�ӱ���������MnO2�У�Mnռ�ȡ�33.3%��HBr35��Hռ��~2.8%��ռ�ȵʹ��������K�п�����һ�����ӻ��������ȫ�µ��ȶ���������Կ���GNoME���ݼ����кܶ����ơ����ӻ���������Ǵ��࣬��һ���ڶ�Ԫ�������м�Ϊͻ����
10. GNoME���ݼ��У����˶�Ԫ�����仯����ռ�ȴ��ӽṹ��ռ��Ҳ�ܴ�����ṹҲ�DZȽ��Ѿ�ȷ�ϳɵġ�ͼ6�п��Կ�����������ռ������Ԫ�صı��������Կ���һЩ����ѧ�ȶ��Ļ������Dz����ࡣ����HBr35����35��Br�в�һ��Hԭ�ӣ�����ʵ���к��ѿ���H������ṹ�������Թ����Ų����γ���Ԥ��һ�µ�HBr35���塣�������ڶ�Ԫ��������ͻ��һЩ������Ԫ����Ԫ��������ռ���������١���ͼ6��
11. �����Ӿ�������ģ���е��Ƚ��㷨�����������ʿ�ѧ��ӵ������֮�ء�ǿ��ѧϰ��ע�������ơ���ɢģ�͡�Ԥѵ��ģ�͡���ģ̬�����������㷨��ģ�Ͷ�����ơ��������ݿ�ȣ����綼��������������Ͽ�ѧ�У���������Ӧ�Ĺ��ߡ�
//
δ�����ص�Զ��������ϣ��
Google��GNoME���ݼ��ǡ�AI+���Ͽ�ѧ�����ʱ�������е�һ������Ȼ���ݼ����������ϸ����û�з����������ɿ��Ա�������δ����������Ļ�ѧ�ռ��У����кܶ�δ֪�²��ϵȴ�����ȥ̽�������ݼ��ķ����������ڿ����˺ܶ�����ԡ�ȫ����о���Ա���л����һ��̽����Щ���ϣ�����Ӧ����Щ���ݴ����������˹�����Ӧ�á����ָ����²��ϡ�����������һ�����ݼ���������һ��չʾ�ſ�������������������µ�·��ͼ��
�ڡ�AI+���Ͽ�ѧ���Ĵ��У�����������֮�ء��������ݼ����ر��Ǿ�����ҵ֧�����õ����ݼ���Ҳ����һ�����������ֺá��Ĺ�������ȴ��һ������ܵġ�Ӳ�̡���
ע�����ľ�����ʽ��Ӣ�İ�����2024��2��28�շ�����Materials Futures��
DOI��10.1088/2752-5724/ad2e0c
URL��https://iopscience.iop.org/article/10.1088/2752-5724/ad2e0c
�����
[1] A. Merchant, S. Batzner, S. S. Schoenholz, M. Aykol, G. Cheon, and E. D. Cubuk, ��Scaling deep learning for materials discovery,�� Nature, vol. 624, no. 7990, pp. 80�C85, Dec. 2023, doi��10.1038/s41586-023-06735-9.
[2] C. Zeni et al., ��MatterGen��a generative model for inorganic materials design,�� Dec. 2023, doi��10.48550/arXiv.2312.03687.
[3] C. Chen et al., ��Accelerating computational materials discovery with artificial intelligence and cloud high-performance computing��from large-scale screening to experimental validation,�� Jan. 2024, [Online]. Available��http://arxiv.org/abs/2401.04070
[4] A. Jain et al., ��Commentary��The materials project��A materials genome approach to accelerating materials innovation,�� APL Materials, vol. 1, no. 1. American Institute of Physics Inc., 2013. doi��10.1063/1.4812323.
[5] J. E. Saal, S. Kirklin, M. Aykol, B. Meredig, and C. Wolverton, ��Materials design and discovery with high-throughput density functional theory��The open quantum materials database��OQMD��,�� JOM, vol. 65, no. 11, pp. 1501�C1509, Nov. 2013, doi��10.1007/s11837-013-0755-4.
[6] B. Deng et al., ��CHGNet as a pretrained universal neural network potential for charge-informed atomistic modelling,�� Nat Mach Intell, vol. 5, no. 9, pp. 1031�C1041, Sep. 2023, doi��10.1038/s42256-023-00716-3.
[7] C. Chen and S. P. Ong, ��A universal graph deep learning interatomic potential for the periodic table,�� Nat Comput Sci, vol. 2, no. 11, pp. 718�C728, Nov. 2022, doi��10.1038/s43588-022-00349-3.