���գ����������ѧ��Harvard University��Eric N. Jacobsen�����鱨���˼�-������-����������4-ȡ������ͪ�����̬��Ҷ���µIJ��Գ�Wittigϩ������Ӧ���ϳ���һϵ�и߶ȶ�ӳ�帻����������ϩ������Ӧ��ʹ�õĴ�����һ�ֺ����Ĵ�����-��-���������������ѧʵ��������������˴�ϩ������Ӧ����Lewis��������Ӷ��ڵ����������γ�����飨oxaphosphetane���Ӻ����������ͨ�����ѽ�����ϩ����������о����������ӳ���ͨ��һ���������еģ����а���������ӳѡ���Եļ���1,2-�ӳɣ��Ӷ�����м�����˼��������سɹ�������J. Am. Chem. Soc.�ϣ���������DOI��10.1021/jacs.4c00564��
��ͼƬ��Դ��J. Am. Chem. Soc.��
����
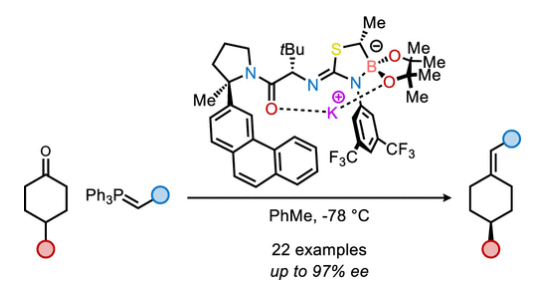
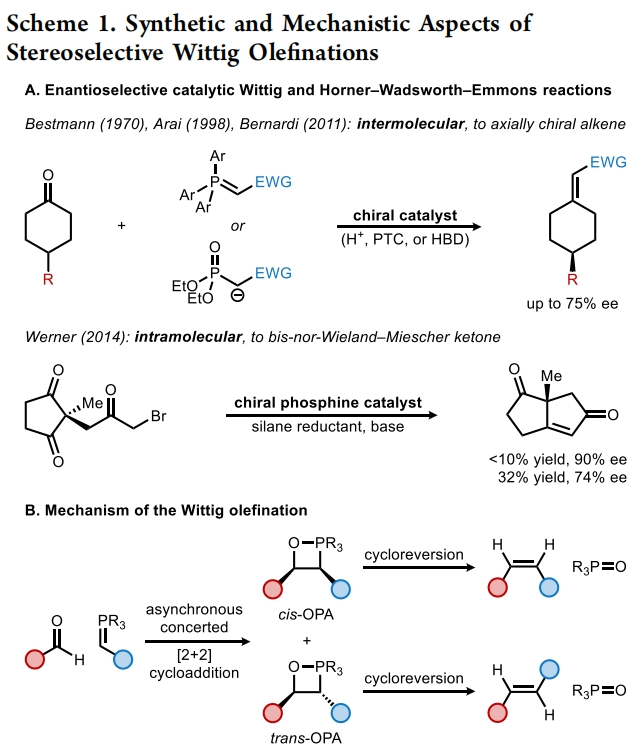
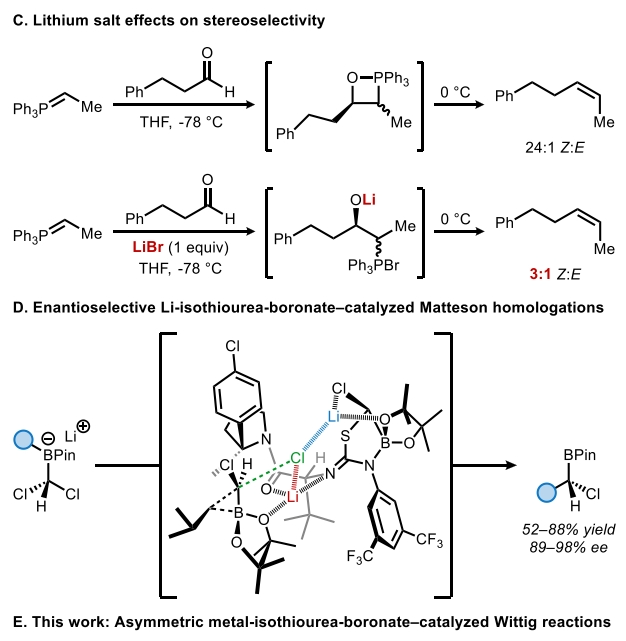
��Wittig��Ӧ�״α�����������㷺Ӧ����ϩ��������ѡ���Ժϳɡ�����ȡ����ǰ����ͪ��ϩ���������γ�������ϩ�����һϵ��ͨ��ʹ�û�ѧ�������Կ������أ����������������������壩��ʵ�ֲ��Գ�Wittig��Horner-Wadsworth-Emmons��Ӧ�ķ����õ��˹㷺��չ���෴��ͨ�����Գƴ���ʵ��Wittig��ϩ�������õ������Բ��������ȴ�dz���������ĿǰΪֹ��������������ͨ��ʹ��Brønsted�ᡢ����������ת�ƴ��������75%��ee�õ������������������ѧEric N. Jacobsen�����鱨���˼�-������-��������4-ȡ������ͪ����ȶ�����Ҷ���µIJ��Գ�Wittigϩ������Ӧ���ϳ���һϵ�и߶ȶ�ӳ�帻����������ϩ������һ���ԵĹؼ��Ƿ���̬Ҷ������������OPAs��oxaphosphetane���ڵ����µ��ȶ��ԣ���Ϊ�ѽ��ϩ����Lewis������좸�����ܿ�����һ��ǿЧ�Ĵ��������Լ���Scheme 1�������ػ�ѧ��APP�����ֻ����ջ������ҵ�������ᡣ
��ͼƬ��Դ��J. Am. Chem. Soc.��
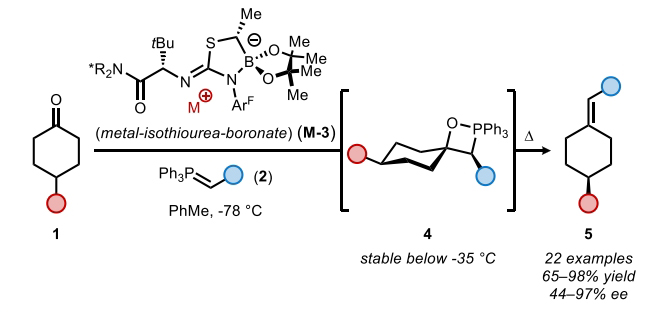
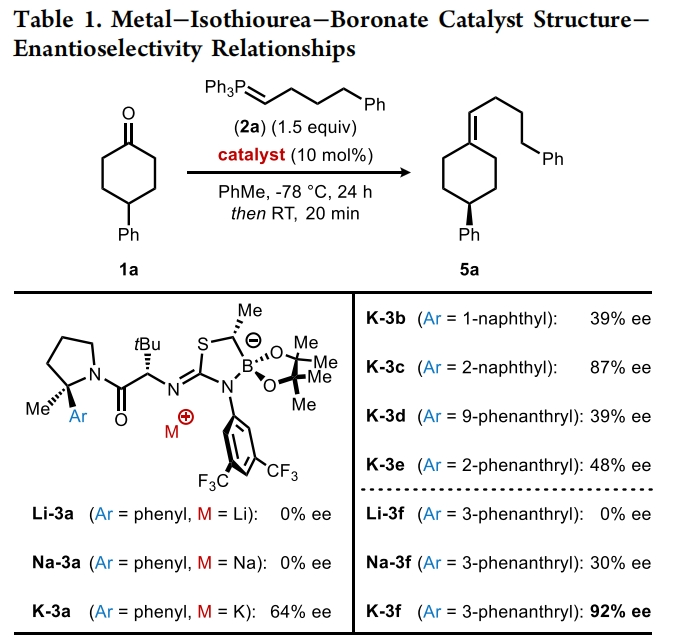
���ȣ�����ʹ��4-��������ͪ1a����Ҷ����2a��Ϊģ�����Է�Ӧ�����������Ż���Table 1������ʹ��1a��1.0 equiv��, 2a��1.5 equiv��, K-3f��10 mol%�����ڼױ���-78 ��C��Ӧ24 Сʱ֮�����·�Ӧ20���ӣ�������73%�IJ��ʣ�92% ee�õ���Ӧ��ϩ������5a��
��ͼƬ��Դ��J. Am. Chem. Soc.��
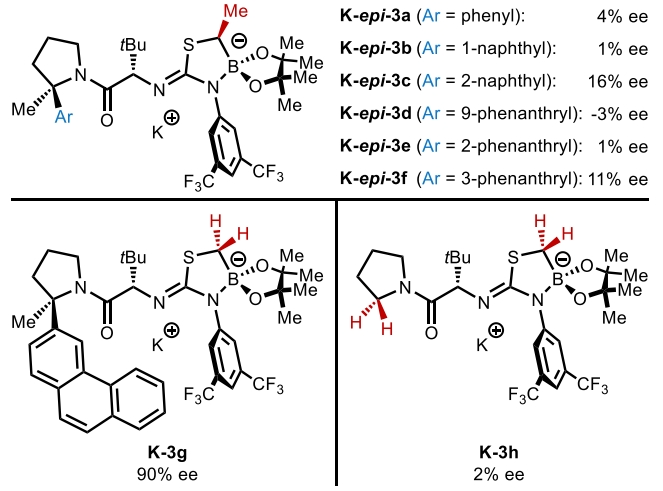
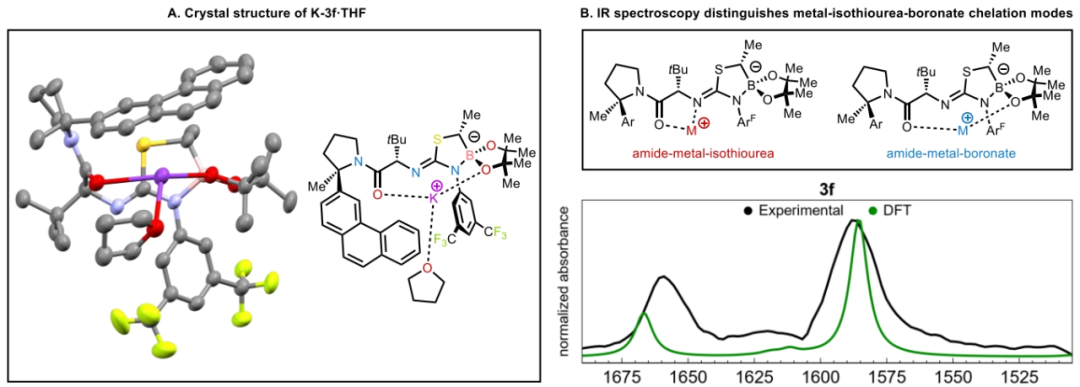
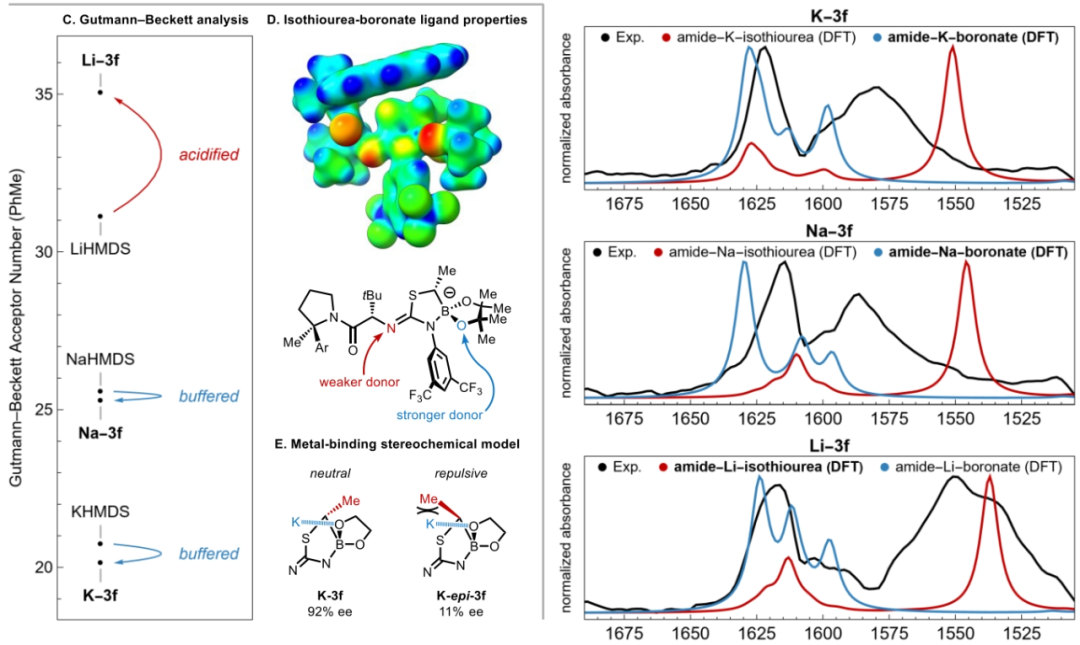
���������ͼ��������ЧӦ�ͼش����ڴ�ת���е���Խ���ܡ�K-3f��THF��X-���߾���ṹ�����ó��������Ϊһ�ֲ�Ѱ���Ĵ���������м���������������������������֮����λ��Figure 1A������������ģʽ��֮ǰ���-������-�������������X-���߽ṹ�����۲쵽����Ԫ����-�-�������������кܴ�IJ�ͬ��Ϊ��������Щ��̬�ṹ�IJ����Լ�����Ӧ��Һ̬�ṹ������ԣ����߲�����M-3f��M = Li, Na, K������Һ������ף���������DFTԤ�������������Ĺ������˱Ƚϣ�Figure 1B����δ��������������-������3f��M = H����ʾ��������C−O��1660 cm−1����������N−C−N��1590 cm−1�����Ӧ������ȣ���Щ������ڼ�������еõ��˺ܺõ����֡����⣬K-3f��Na-3f�ĺ���������ʻ�������ʾ�����Ƶ�ģʽ��C−O����ĺ��ƴ�ԼΪ40 cm−1����N−C−N����ĺ��������3f�Ĺ��׳ʱ�ƽ״����Щ���ױ仯��Ԥ�������-����-������������ĺ��������һ�¡����֮�£�Li-3f�ĺ��������ʾ������ĺ��ƴ�ԼΪ40 cm−1������������-����-��������������һ�¡���Щ����ģʽ�Ļ����������ת��Ϊ���Lewis���ԵIJ��죬��Gutmann-Beckett������ʾ��Figure 1C�����ֽ����ó�K-3f�����KHMDS��Na-3f�����NaHMDS��Lewis�������в�ͬ����Li-3f�����LiHMDS��Lewis�����������ӡ�����3f������������-�����������ӵľ����λͼ�У�Figure 1D��������������ԭ�Ӵ������˽ϸߵĵ����ܶȣ��������������������-����-�����������������۲쵽��Lewis���ԵĻ�����һ�¡�ʹ��K-epi-3a−f�����ĵͶ�ӳѡ���ԣ�Table 1���ɹ�����˳ʽ ��-���������ЧӦ������ܻ��������-K-��������������γɣ�Figure 1E����

��ͼƬ��Դ��J. Am. Chem. Soc.��
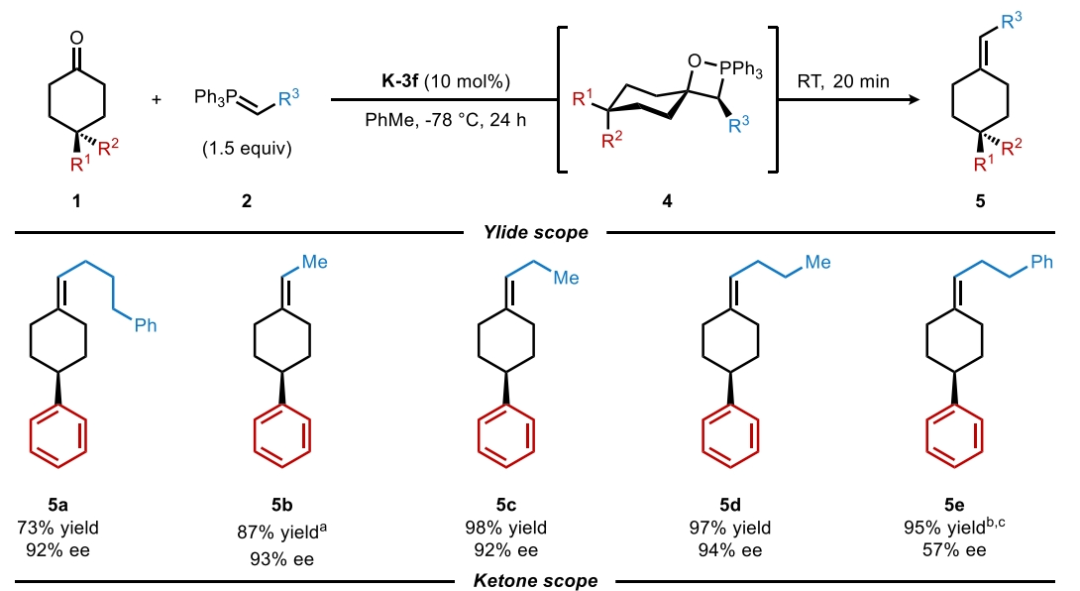
�ڵõ������ŷ�Ӧ���������߶Դ�ת���ĵ��ﷶΧ������̽����ʵ��������һϵ�в�ͬȡ���Ļ���ͪ1����Ҷ����2���������õļ����ԣ���65-98%�IJ��ʣ�44-97% ee�õ���Ӧ��ϩ������5a-5o��Figure 2�������а��������������������������������ԭ�ӡ��嶡�����������һϵ�л����ڷ�Ӧ�о��������õ������ԡ�
��ͼƬ��Դ��J. Am. Chem. Soc.��
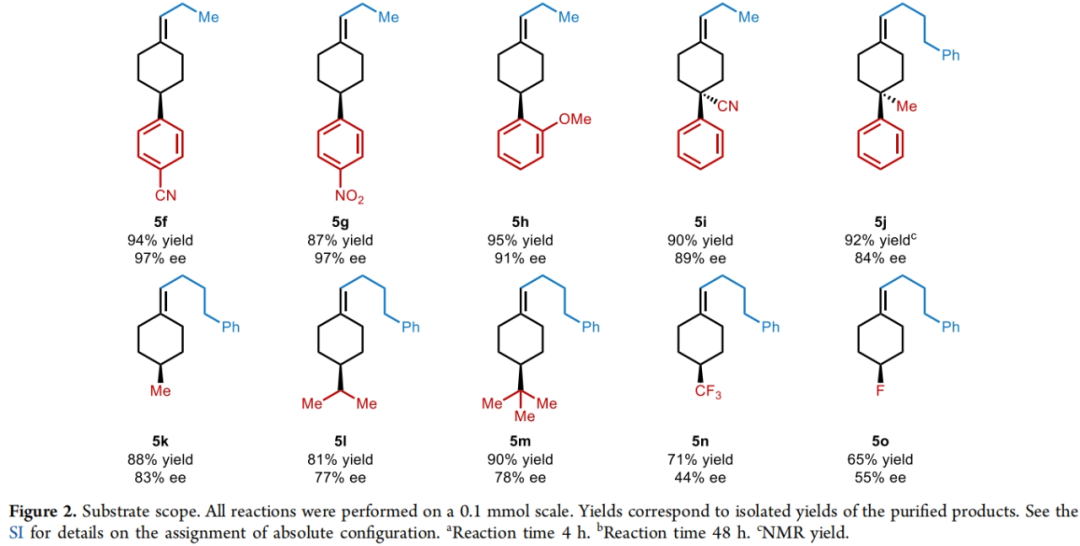
������������̽���˵���ЧӦ��ee��Ӱ�죨Figure 3����һϵ�в�ͬȡ����4-��������ͪ����ķǴ�ϩ������Ӧ�л���������γ�C-C���ĵ糡ǿ���кܺõĶ�Ӧ��ϵ��Figure 3A�������⣬���߶�һ�鲻ͬȡ����4-��������ͪ��k-3f��ϩ����Ӧ�����о��������ﺬ��ȱ���ӷ���ʱ����ӳѡ���Ը��ߣ�Figure 3B����������߽�����G⧧�뼸����ͬ�ı����������˶Աȣ��������meta���������ã�Figure 3C�������⣬�����ӻ��Է�Ӧ����Ҳ������Ӱ�죬�ڴ��ͷǴ���ϩ������Ӧ�У���λ��������ȡ�������ܼӿ췴Ӧ�ٶȣ�Figure 3D����
��ͼƬ��Դ��J. Am. Chem. Soc.��
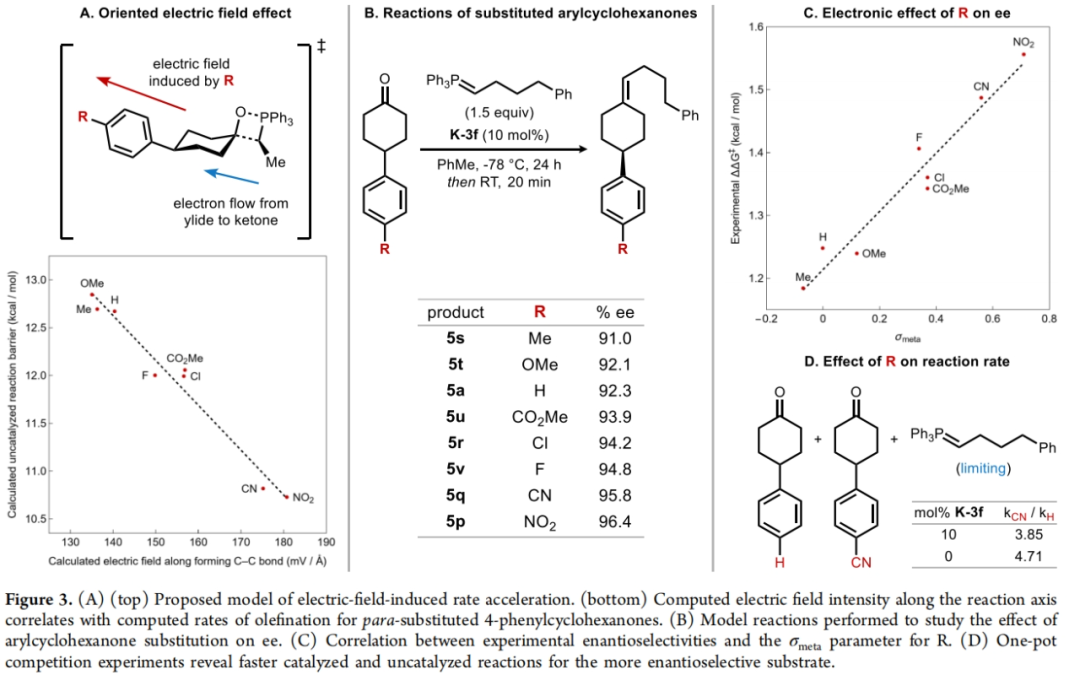
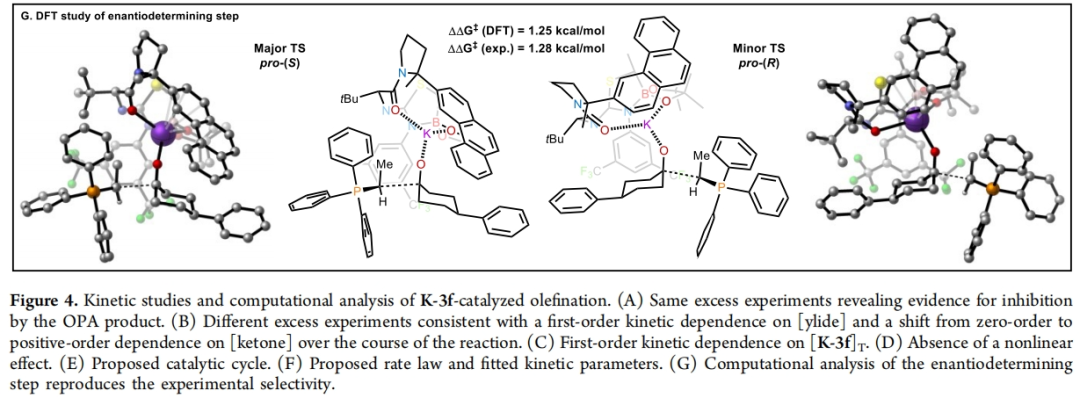
���������������-�ɼ�����ͪ1a��ϩ�������̽����������Ķ���ѧ��������������520 nm���Ⱥ�ɫ��Ҷ���� 2d����ʧ�����⣬����Ũ�ȵı仯��ʾ��һ������ѧЧӦ������䲻���ڷ�����ЧӦ������������ѭ���д��ڵ����������һ�£�Figure 4A-4D������������ʵ��������������˿��ܵķ�Ӧ������Figure 4E��������K-3f������ͪ1a����γ������A����������Ҷ����2d�IJ�����ӳ��γ���˼������B�����������Կ���Ľ���OPA����4d���õ������K-3f�����⣬DFT����ó�����״̬������״��Ҫ���������ؾ�����Figure 4G����1������ͪ�ӳɵ���ѡ���Ժͣ�2��������좲���������Ǽ�֮��Ŀռ��������С�����������Ҷ����ȡ�����ķ���
��ͼƬ��Դ��J. Am. Chem. Soc.��
�ܽ�
Eric N. Jacobsen���������þ������ʹ����Ͻṹ������Lewis���-������-��������������ʵ����4-ȡ������ͪ�ĸ߶�ӳѡ����Wittigϩ������Ӧ��������ӳѡ���ԵĻ��ӳɹ��̰���������ļ���1,2-�ӳ��γ���˼�����������п��滷���ķֲ����̡��˷�Ӧ�ķ�չΪ�����Գ�Wittigϩ�����ķ�չ�ṩ���µ�;����