����
���գ�2021��ŵ������ѧ������������˹�ٴ�ѧĬ�˴�����David W. C. MacMillan���ڿ�������Science�ϱ�����һ�������ɻ���ѡ�鵼�Ĵ�-������ż��������������Ƭ����ͬһ����Ӧ����������ż�����Կ������š�Ruizhe Chen��Nicholas E. Intermaggio��л������ʿ��James A. Rossi-AshtonΪ���Ĺ�ͬ��һ���ߣ���������DOI��10.1126/science.adl5890
����
�л���ѧ��һ���ؼ�Ŀ���Ƿ����ܹ��ӷḻ���ȶ�����ʼԭ���γ���C-C���ĺϳɷ���������һ�ָ���C��sp3������Ȼ�����ţ�����Ȼ����ҵ��Դ���ձ���ڡ�������Ƭ��֮��ĵ������Ž���ż�����ܹ����ǰ��δ�еĻ�ѧ�ռ䣨ͼ1A����
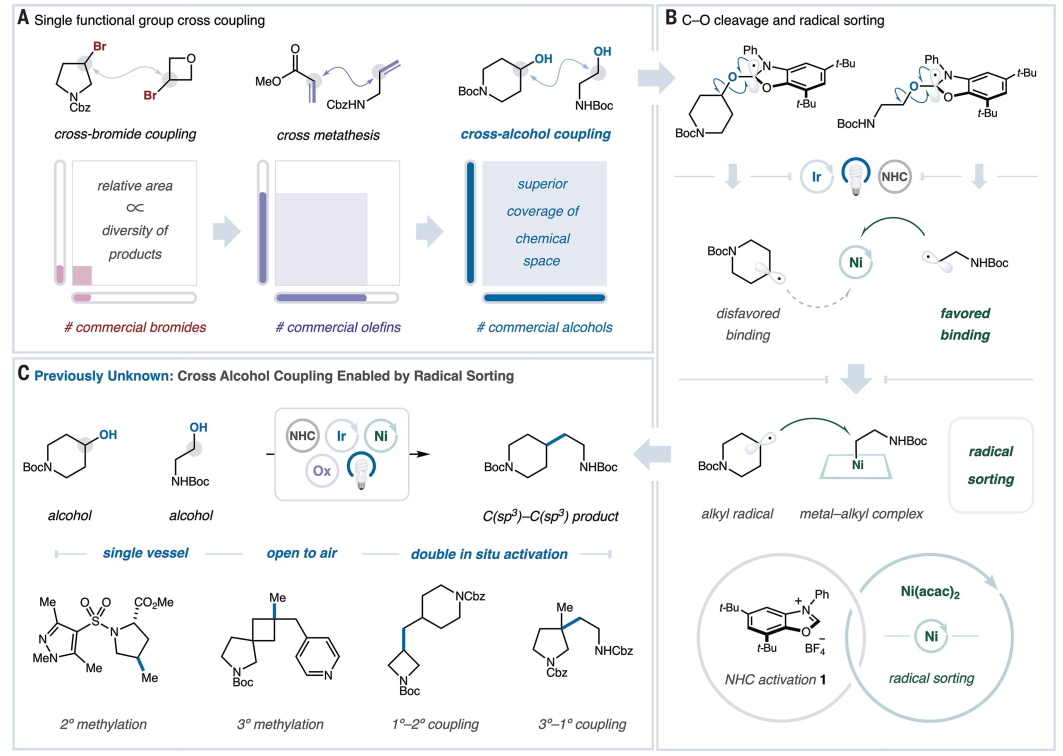
ͼ1. ��Ӧ��ƣ�ͼƬ��Դ��Science��
�������������ʹ��N-�ӻ�������NHC���ζԴ����й�������ԭԭλ������Nature, 2021, 598, 451-456������ƽ̨�����ڼ���һ�ִ��ṹ��Ԫ������ת��Ϊ˲̬������ɻ���Ȼ���ܹ��������ż���γ��¼���Ԥ����ЩNHC�Լ�������ͬʱ����ͬһ��ƿ�ڵ����������飬Ϊ�߶�ģ�黯�Ľ��洼ż���ṩ���������⣬���۴�ȡ��ģʽ��Σ�NHC-���Ӻ��ﶼ����ٴ��������ļ���̬�����ֹ��һ��ż����ż���������һ��ż����ż����������ġ�������ˣ���Ӧ;����ͨ�����ֲ�ͬ���ɻ����н���У��������ͨ���ᵼ��������绯����ĸ��ӻ������ػ�ѧ��APP�����ֻ����ջ������ҵ�������ᡣ
Ϊ��Ӧ����һ��ս��������ͼ���ù��ɽ����ڿռ�������ȶ�������������ɻ�������������-�������ǿ���������ȡ���ȵ����Ӷ����͡��������Ե��¡����ɻ���ѡ��ЧӦ�������������ɻ���ȡ���̶Ƚϵ͵ı�ѡ���Բ��Ӷ��γɸ��ȶ��Ľ���-����������⣬���ʵĽ��������������з�ѡ���ɻ���DZ�������һ�����ͨ��˫���Ӿ���ȡ����SH2���鵼C��sp3��-C��sp3�������γɡ����ɻ���ѡ��SH2�Ľ�Ͽ��Գɹ��鵼����˲ʱ������ɻ�֮��Ľ���ѡ����C��sp3��-C��sp3��ż����ͼ1B����
��Ӧ������ͼ2��ʾ�����ִ����뱽���f���f1��NHC-1��Ԥ��ϣ�ͬʱ�ڵ�����Ӧ�������γ����� NHC-���Ӻ�������һ�Ӻ�������ʹ�����ļ���̬��Ͻ�����ͨ������-ȥ���ӻ��ͦ�-�����γ���Ӧ��������ɻ����º͵����������Կ���ת�仹ԭ�Ĺ������ʹ��ָ���Ir��III����̬�������ڶ��ι����������������������ɻ���һ���γɣ�������ɻ���ͨ�����ʵĽ����������з��࣬��������C��sp3��-C��sp3������ż����
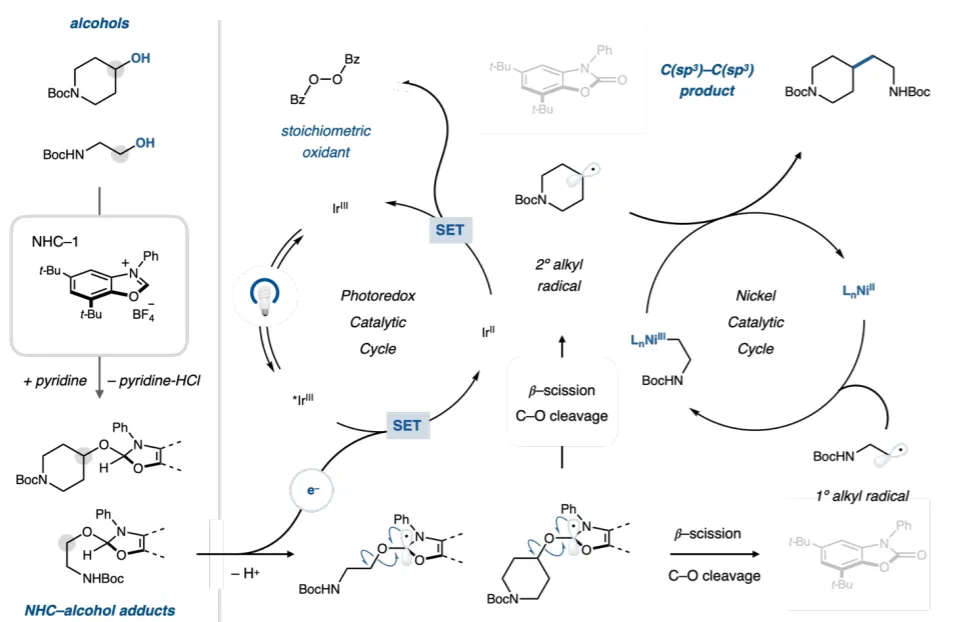
ͼ2. ���ܵķ�Ӧ���ƣ�ͼƬ��Դ��Science��
���ȣ����߽��Ż����ص�����٣�2�㣩����״��Ľ���ż���ϡ�ֻ���������������½����ִ�������NHC��1��ϣ�Ȼ��ֱ������ԭλ���ɵ�SH2���� Tp*Ni��acac����Ir[dF��CF3��ppy]2��dtbbpy��PF6�����������������������Ͷ���������DMSO������ɫ��������ܣ�LED������1Сʱ������߲��ʵļ���������ɻ���ѡ��������Ч�ģ���������ɻ�-���ɻ�������ȣ�ʹ������˲ʱ������ɻ��ܹ��Ը��ߵ�Ч�ʽ�ϡ��ڱ��ĵ�ת���У�����Ҫ����f�ι��ˣ���ɼ�Ӧװ�ã����д��ڵ�����Ӧ��ƿ�б���������ͽ���ż����������Ҫ���ǣ������ȡ�κ�Ԥ����ʩ���ų�������ˮ�ּ��ɱ��ַ�ӦЧ�ʣ���һ����ͻ���˸÷������Ƚ��Ժ��û��Ѻ��ԡ����ĵĽ��洼ż��ͨ������Ҫ1.0��1.5�����ļ״�����ʵ�ָ߲��ʡ�
������ѷ�Ӧ����������̽��������״��Ľ��洼ż���ķ�Χ��ͼ 3�����������ɻ�ȡ��ģʽ�����С��������������״���Ч����ż���������������� 2������ 51%���� 3������ 55%���������ٴ������ֻ�ϵ��������Ԫ����Ԫ����Ԫ����Ԫ�����ӻ���������������������IJ��ʣ�4-11��55��70%���ʣ����м�����һϵ���ݻ���ϵҲ�����õIJ��ʽ�������Ч�Ľ���ż����12-15��51%��63%�IJ��ʣ������⣬ͨ���ؽ��״���CD3OD �����������ٴ�L-�հ������ֱ��ת��Ϊ�dz����L-�Ӱ���-d3������ 66%������ʹ���߳��Ĵ�ͷ�ϳɡ����⣬�߱��״��ͱ�����ϵͳ���ӵĴ�����ֱ�Ӽ��������ʷֱ�Ϊ 56% �� 68%��18 �� 19����
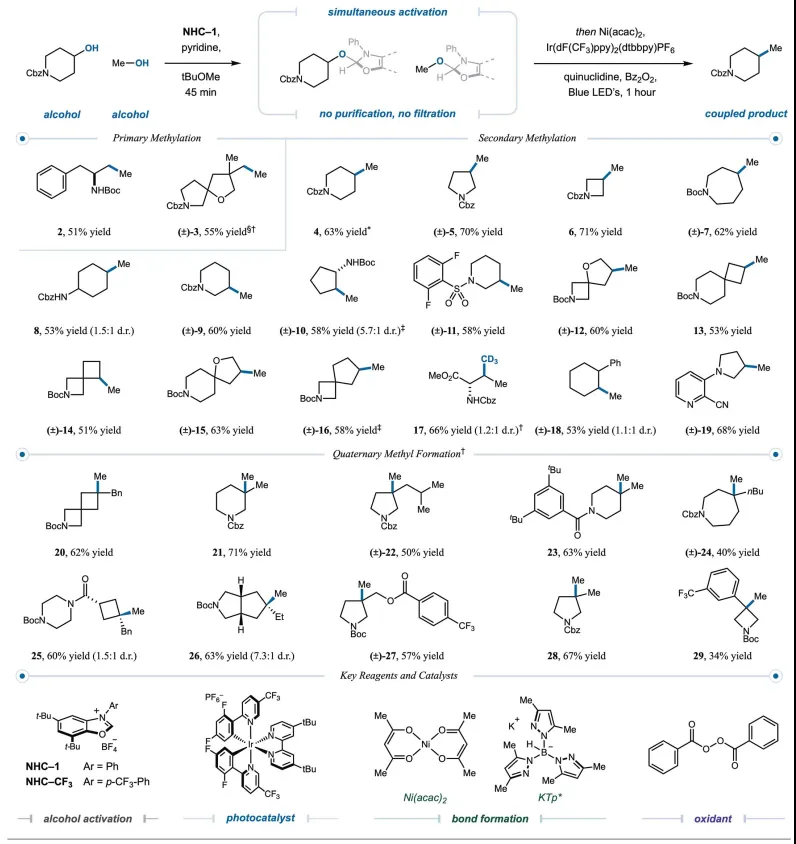
ͼ3. �ͼ״����洼ż���ĵ��ﷶΧ��ͼƬ��Դ��Science��
����ʹ�����Ը�ǿ����������NHC�����NHC-CF3����2�����ļ״�����һϵ���崼������һ�������ķ�Ӧ���������ֿ��Թ���һϵ�нṹ�����ļ�̼��
��������Ԫ����Ԫ����Ԫ����Ԫ�����ӻ��ϵĻ�״�崼ȫ��ת��Ϊ�ϳ������õļ���������������죨21-25��28��40%��71%���ʣ������⣬�����崼���ݻ���˫����ϵҲ���Խ���ż�����ֱ���62%�IJ��ʣ�20����63%�IJ��ʣ�26����������������崼���ڵ�����ԭ�Ӿ������õ������ԣ��Ӷ��������õļ�̼�γɲ��ʣ�27������57%���������ͨ����״��Ľ��洼ż����������������̼����ϳɲ��ʣ�29��34%����
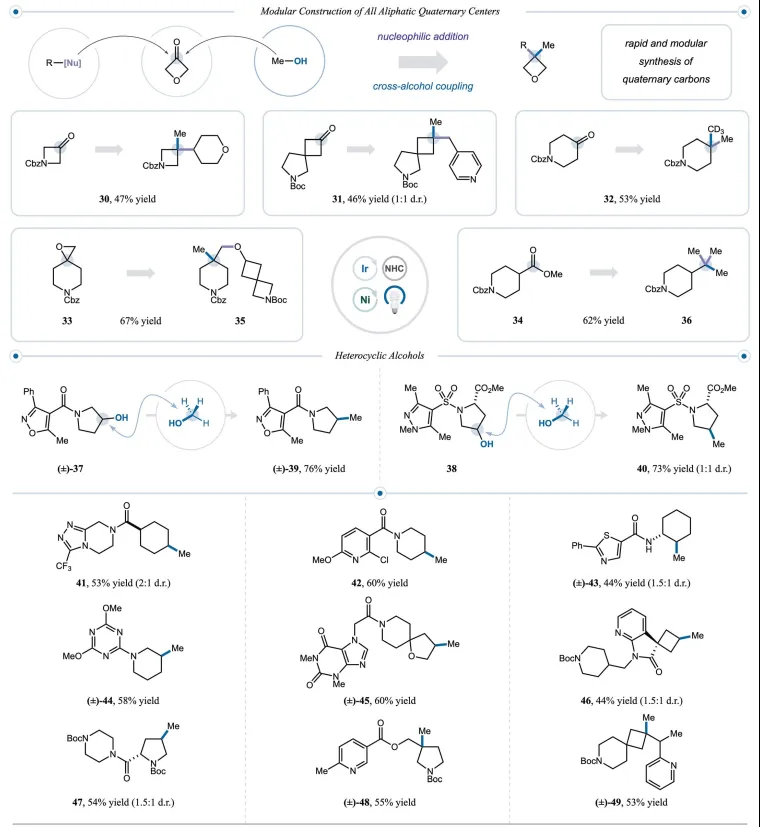
ͼ4. ȫ̼��̼���ĺ��ӻ�����ģ�黯������ͼƬ��Դ��Science��
�����崼����ͨ�����˼ӳ�����Ȼ������Ź㷺��ã�������ʶ���л���ͨ�������ϳɲ��轫ͪ�����ͻ�������ת��Ϊ��̼��ͼ4����Ϊ�ˣ���һϵ��ͪ��ṹ��ͬ���л��������Լ���Ӧ��������ת��Ϊ��Ӧ��3�㴼����Щ�����Բ�����״���CD3OD�Ľ��洼ż���������õIJ����ṩ��̼30��31��32 �����ʷֱ�Ϊ 47%��47%��53%�������⣬�Ի�������33�ͼ���34�����˼ӳɣ�Ȼ�������崼�����������õIJ����ṩ�˼�̼����35��36�����ʷֱ�Ϊ67%��62%����
���������߽�ע����ת�����ҩ������ӻ��Ĵ��ļ�����Ϊ�ˣ�����f��37�����������38���ڱ��ĵķ�Ӧ�����£�������IJ�������������ļ���������⣬�������41������43�����44�����47������ת��Ϊ�ϳ������õ���Ӧ��������������ã�44%��58%���ʣ���
�ӻ����������� 42������ 45 �͵������� 46�����ܶԾ����������ǽ���ż��������ս����Ϊ������ Minisci �ͷ�Ӧ�������ڽ������ɻ���Ȼ�����ڱ��ĵ���������£��������д����ӻ��Ĵ������õIJ��ʣ�43����60���IJ��ʣ����м��������⣬�����48��49�γɼ�̼���ģ����ʷֱ�Ϊ 55%��53%��
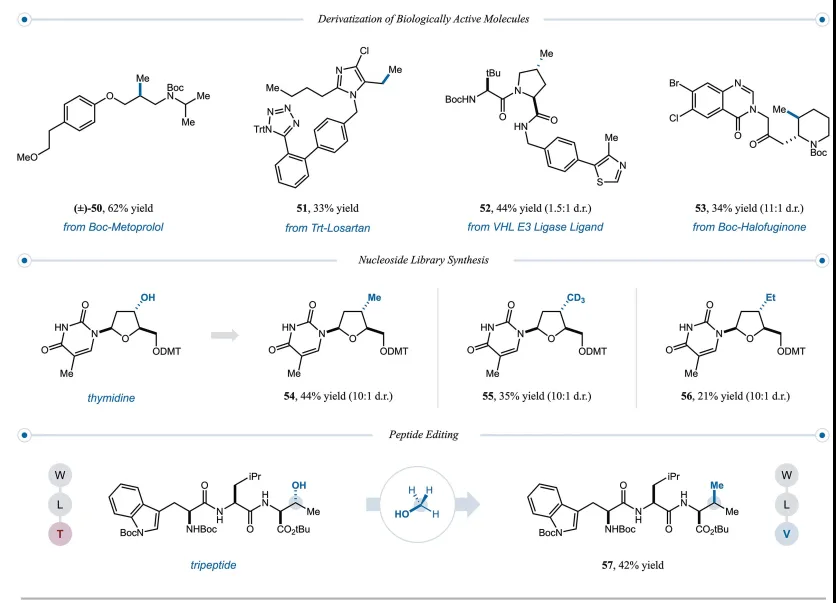
ͼ5. ���ӷ���Ӧ�ã�ͼƬ��Դ��Science��
����̽����������Է��ӵĺ��ڹ��ܻ���ͼ5�������������50������ɳ̹��51����VHL E3����ø��ϼ���52����±��ͪ��53���ļ��������ﶼ�����ںϳɺ��ںϳɣ��������õIJ��ʣ�33%��62%���ʣ���������ͨ���ಽ�ϳ���Щ�����������ʱ�����Դ��
��������״���CD3OD���Ҵ����н��洼ż��������ļ���CD3���һ�������IJ��ʷֱ�Ϊ44%��54����35%��55���� 21%��56����
��������ں��������εı�����̽���˽��洼ż������ɫ����-������-�հ����������ڽ��洼���������¡��հ�������е����ǻ�����Ľ��������ɰ��������Ӱ���л��Ļ��䡣�Ӱ������57�IJ���Ϊ42%������ͨ��һ���ĺϳɲ����ɹ��ؽ����Ĵ�W-L-T �༭ΪW-L-V���ڳ���̽���˸��ִ��ͼ״�֮��Ľ��洼ż�������߽�����̽����ͨ���������ֲ�ͬȡ���Ĵ�֮��Ľ���ż���γ�һ���C��sp3��-C��sp3������
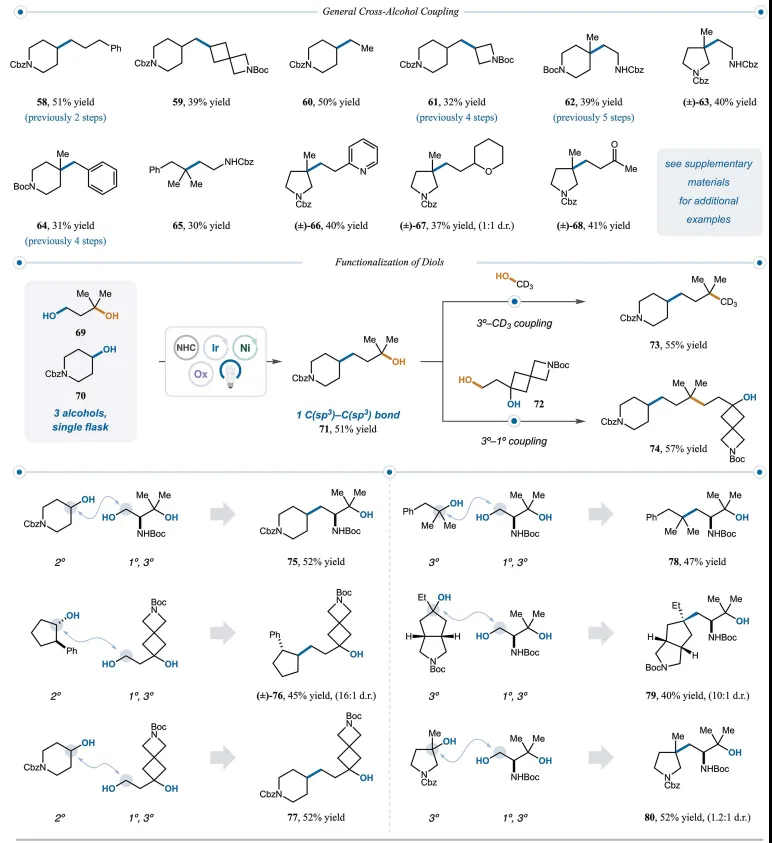
ͼ6. һ�㽻�洼ż����ͼƬ��Դ��Science��
����ѷ�Ӧ�����£���������ʵ�ָ߶�ģ�黯��C��sp3��-C��sp3��Ƭ��ż����ͼ6������ˣ���ͬ���ߴ�� 2�� �� 1�� ���ںϳ��Ͼ������õ�Ч�ʣ�58-61��32%��51% ���ʣ���ֵ��ע����ǣ�3���1�㴼֮��Ľ��洼ż��������ģ�黯���û��Ѻõķ�ʽ������̼����״�����崼��һ�κϳɲ�����ż�����Ժϳ����õ����ʣ�62- 68��30%��41%���ʣ��ṩȫ֬���弾̼���
�������������о��˶����ĵ������ܻ���Ϊ���ٻ�ø��ӵġ�����C��sp3���ṹ�ķ���������NHC��֮������������ɿռ�λ�������MeOH > 1�� > 2�� >> 3�㣩��������һ���ƣ�Ԥ�Ʋ���ȡ���Ķ�������������ż�������������ֱ�����1�㡢3�� ������69������2�㴼70��ͬ����ƿ�н������ϡ�����ͨ�� 1�� �� 2�� ����ѡ������������51%�IJ����γ���71���ڴ˽Σ�71��ʣ���3�㴼������CD3OD��72������һ�ֽ��洼ż�����ֱ���55%��57%�IJ����ṩ���ӵļ�̼����73��74����ʵ֤�������ֵ������ܻ��ĵ�һ���dz��Ƚ�����ʵ�ָ�ԭ�������������β������� 700 ���˵��м��崼 71��1�㡢3������Ĺ����Ż��dz��ɹ����ṩ��һϵ�и��ӵ�2��-1��ż�����75-77��45%��52%���ʣ��Լ�3��-1��ż�����78-80��40%��52%���ʣ���
�ܽ�
David W. C. MacMillan�Ŷӱ�����һ�ֽ���ż���������ü���ͨ����һ���Ƚ����û��ѺõIJ������ḻ�Ͷ����������Ƭ�ι������ֲ�ͬ����C��sp3��-C��sp3���������ĵĽ��洼ż����������Ӧ�û�ѧ��ѧ����ֱ��Ӱ�죬���ḻ��̼̼���Ĺ���������
�������飺
Ruizhe Chen, Nicholas E. Intermaggio, Jiaxin Xie, James A. Rossi-Ashton, Colin A. Gould, Robert T. Martin, Jes��s Alc��zar, David W. C. MacMillan*. Alcohol-alcohol cross-coupling enabled by SH2 radical sorting. Science, 2024, https://www.science.org/doi/10.1126/science.adl5890