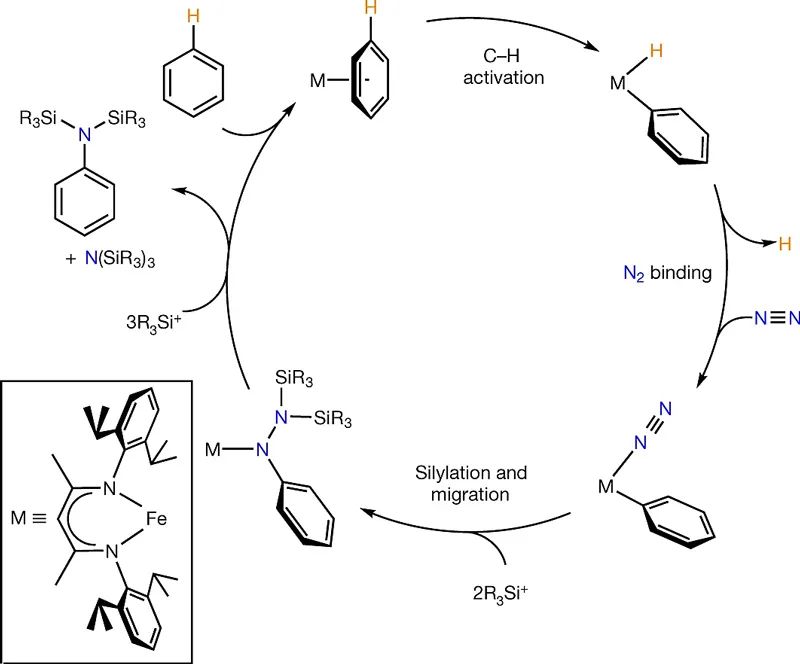
��������N2����Ҷ���İ����Сѧ��ѧ������Ѿ����������Ǻ����Ŀ�����Լ78%����������˷ḻ�ĺ�����ʹ�õ�����Ϊ�Ʊ����������������ԭ�ϡ����������������Թѵ�������N2�����е�N��N��������Ȼ������ǿ�Ļ�ѧ��֮һ����Ҫ˳���N2һֱ���ľ���ս����ũҵ����Ҫ����൪�ʵ�ҩ���г����ĵ��ӻ����������������������ķ������涼�dz���Ҫ����˹̵�һֱ�ǻ�ѧ�����һ����Ҫ�о����⡣��Ȼ��ѧ���Ѿ������������ܰѵ���ֱ�ӻ�ԭΪ���Ĵ���ϵ������ͨ���̵����γ�̼������C-N��ȴ��̫���ס���ͳ��������N2��Ϊ��Դ���ϳɺ����л��һ��Ҫ�ڻ�ԭ�������°�N2ת��Ϊ�����Լ���Ȼ����̼���Լ���Ӧ���γ�Ŀ��C-N�������ǣ�ǿ��ԭ�������£�̼���Լ�����Ҳ�ᱻ��ԭ��ʹ�øò����ڴ��������²������С���Դ����⣬������һֱΧ�����������������չ���о�������Ү³��ѧ��Patrick L. Holland���ڿ����������ܷ�ֱ���յ�N2������γ�C�CN�����������������Ԥ�ȹ����Ż����罫��ת��Ϊ���Լ��������գ�������Nature �ϱ������������˳������N2���Ʊ���������������̵���Ӧ������������ԣ�ͨ��C-H������������Ա���C-H�����������ӳɣ��γɺ�����-̼�����м��壻�����м�����N2��ϣ��ں���Lewis�������£�����N2�����Դ����ǿ��ʹ�ñ������Է���Ǩ�Ʒ�Ӧ�γ����м��壻�ڻ�ԭ�������£��µ�N-N�����ѣ����ɱ��������ͬʱ�ڹ���Lewis��������£��ͷų���������������һ����ѭ����ͼ1����
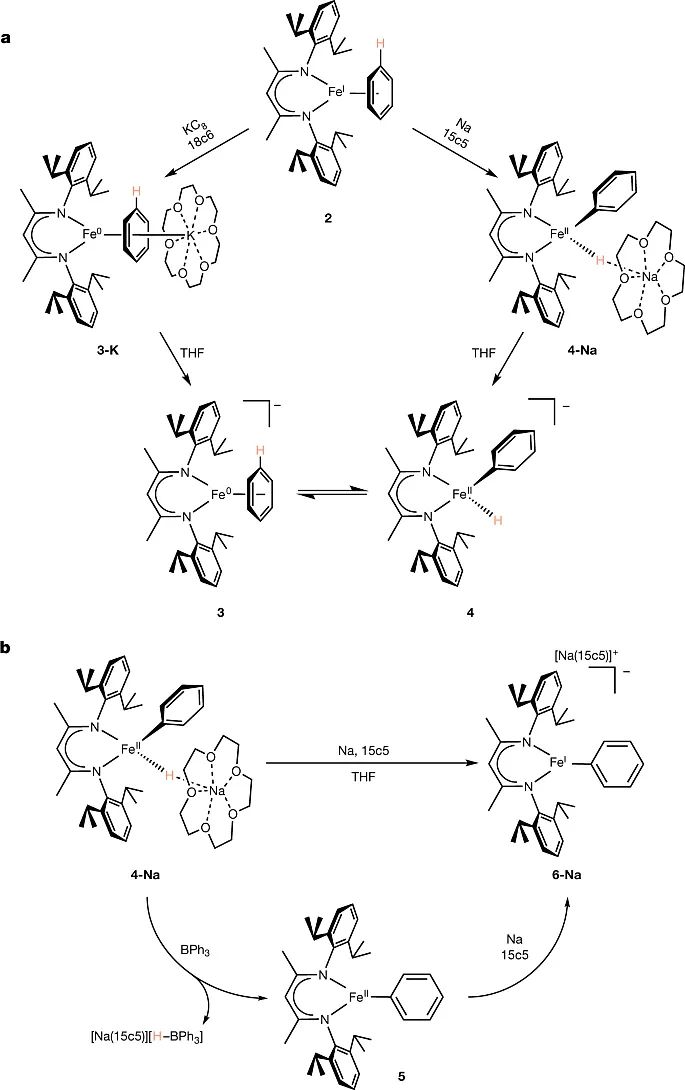
Ϊ���о��ò��ԵĿ����ԣ��������Ƚ����˵�����Ӧ�о���ͼ2�������ȣ����Ǻϳ�����-�������LFe����6-C6H6����2����Ȼ��ֱ��ڲ�ͬ�Ļ�ԭ����������ԭ��������KC8��18-��-6��18c6�������£������2���Ա���ԭΪ����������LFe����4-C6H6��K��18c6����3-K���������ƺ�15-��-5��15c5�������£������2��ԭΪ�����������LFe��H����Ph��Na��15c5����4-Na����ͨ������3-K��4-Na ����˹������Mössbauer�����ܶȷ������ۼ��㣬������3-K��4-Na���в�ͬ����ԭ�ӳɼ���������4-Na�б����ϵ�һ��C-H�����ѣ��γ���һ��̼-��������-��������3-K�����DZ����Ħе�������ԭ����λ�����⣬��뮴��ܼ�C6D6�£����߶�3-K��4-Na���ȶ��Խ����˺˴���ʵ�顣�����ʾ����Сʱ����������һЩ�˴ŷ���ʧ�ˣ���������Щ�������ı�����뮴��ܼ�C6D6�����˽�����ͬʱ��3-K�ĺ˴���ͼ�г�����4-Na�ķ壬��4-Na�ĺ˴���ͼ��Ҳ������3-K�ķ壬����������3��4֮����ڻ�ѧƽ�⡣�û�ѧƽ��֤ʵ�����Ա���C-H���������ӳ��ǿ���ģ�ͼ2a��������4-Na��THF��Һ�����ӽ����ƺ�15c5ʱ���������¾��ü�Сʱ��4-Na����ɫ��Һ���������̡��˴�����֤����һ���������[LFePh][Na��15c5��]��6-Na�������ɡ�������ǿLewis��BPh3ʱ��4-Na�е��⸺������ת�Ƶ�BPh3�ϣ�������֪������5��[Na��15c5��][H-BPh3]����5���ձ�3�����ƻ�ԭΪ6-Na��ͼ2b����
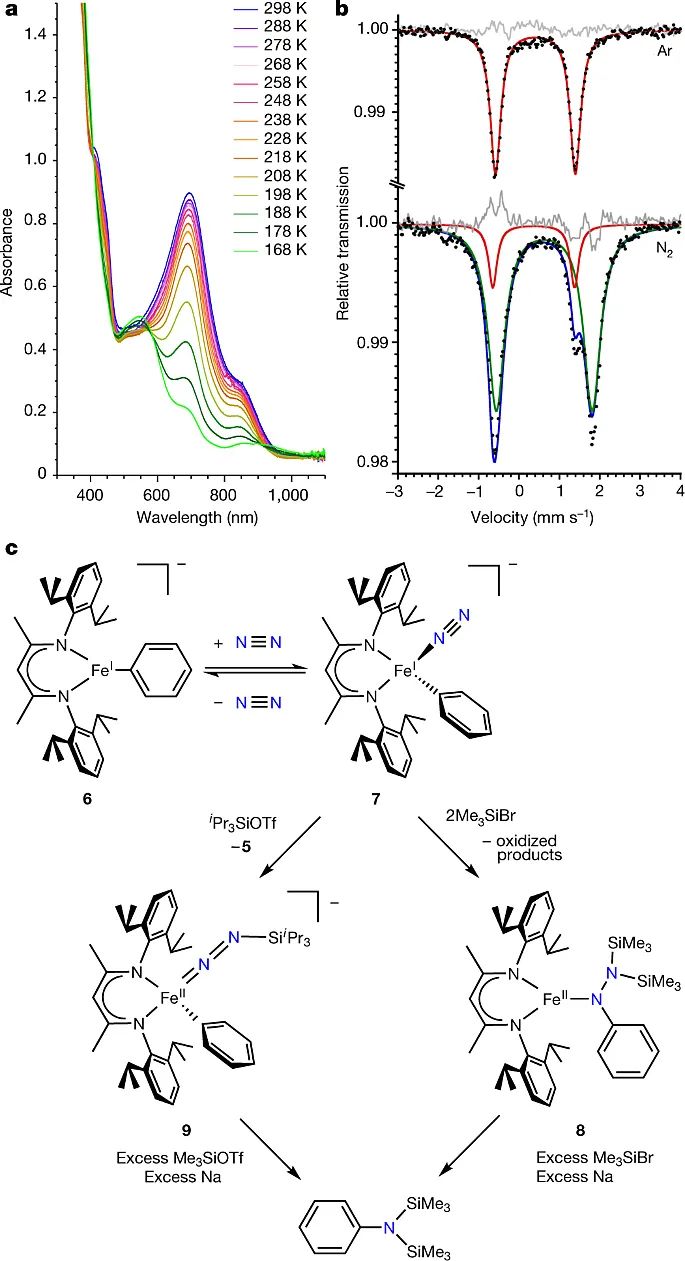
���о��˱����Ļ�����߰�Ŀ��ת����N2�Ļ��ͼ3�������ȣ�������N2�����¸ı��¶ȣ�����6-Na������ɼ����գ�UV-vis������ͼ����˹�������ᷢ���仯������Ar�����£���Ӧ����ͼ��û�з����ı䣨ͼ3a��3b����������������6-Na������N2��ϡ����ţ����߽����˾���ѧ��֤������-78 ��C�½���������6-K��ȴ3 h���õ�N2�����7-K����X-���߾���������ʾN2��һ����ԭ�ӹ¶Ե�������ԭ����λ�����������Ϊ�����幹�ͣ�ͼ4��������������������7-K��Һ�������˲�ͬ�Ĺ��Lewis�ᡣ��ʹ��������Me3SiBrʱ�������7-Kֱ�ӱ�ת��Ϊ�������LFe��N��Ph��N��SiMe3��2����8���������������ӽ�������Ǩ�Ƶ�N2�Ħ�-λ�������γɼ�����������Ĺؼ�֮����������8�����ӹ�����Me3SiBr�ͻ�ԭ����ʱ��8��ԭΪ������������ı����������7-K������λ������������������������Σ�TIPSOTf���������7-K��ת��Ϊ�м���9���侧��ṹ��ʾ������δǨ�ƣ�������ԭ������Lewis�����λ�����ڱ���Ǩ��֮ǰ��ͼ3c�������ţ���9�����ӹ�����Lewis��Me3SiOTf�ͻ�ԭ���ƣ�9Ҳ�ᱻ��ԭΪ�������������14%��
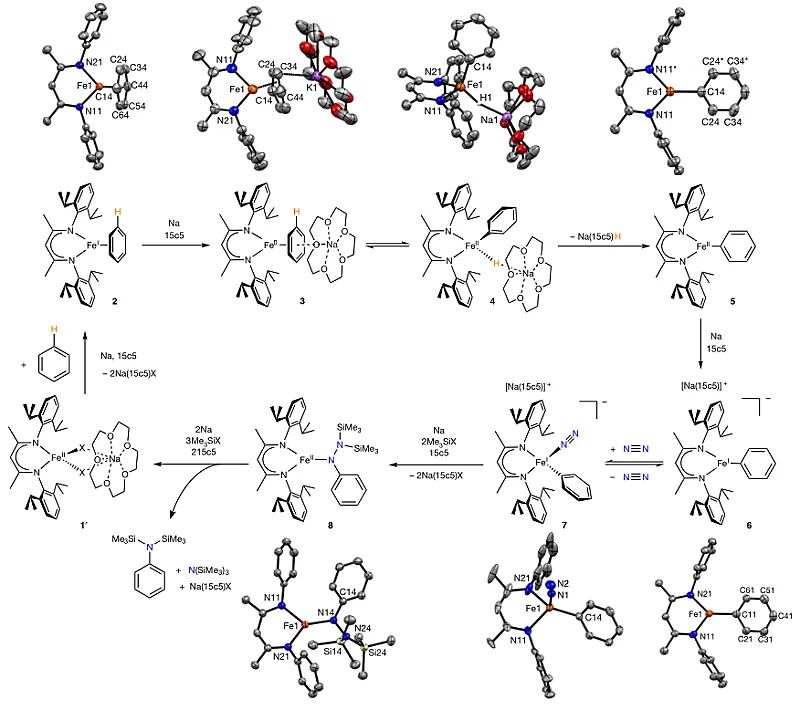
���ˣ���Ȼ�����Ѿ��õ���Ŀ����ﱽ����������Щ���ݲ�����˵���ù�����һ����ѭ���Ĺ��̡�Ϊ�˻ش�����⣬�������м���8����Һ�������˼ױ�����Ϊ����ķ�Ӧԭ��Ϊ����������Ӽױ���Ҳ�ܵõ����ڼױ��İ����������֤ʵ���ڵ�һ�ַ�Ӧ�����������Ȼ���з�Ӧ���ԣ����Լ���������һ��ѭ�����Ӷ����ϴ��Ķ��塣ʵ���������������Ӽױ��õ��˻��ڼױ��İ�������Ӷ�֤���˸÷�Ӧ��һ�������̡��ڴ˻����ϣ�������������µĴ�ѭ����ͼ4�������ȣ������1'���ƻ�ԭ�������뱽��Ӧ����һ�����м���2�����߿��Ա��������ƽ�һ����ԭΪ������м���3��ͨ��������м���Ա���C-H���������ӳɣ�3��ת��Ϊ�м���4�����ʧȥ�⸺���������м���5�����߽�һ������ԭΪһ�����м���6�����ͬʱ������һ��������N2��ϵĿ�λ������N2��������м���7����ǿLewis�������£�N��N����������м���7�ϵı�������Ǩ�ƽ��������м���8���ڻ�ԭ�������£��м���8�е�N-N������ԭ�Ӷ�����Ŀ��������������������1'�������������ѭ����
��ȷ����ѭ�������߶�������ʼԭ�ϱ�����N2������ͬλ�ر��ʵ�飨ͼ5��������15N2�����½��з�Ӧʱ������ɫ��-��������ʾ��Ӧ���ɵı���������Ph15N��SiMe3��2�к���15N��������N2�Dz���Nԭ�ӵ���Դ��ͬ������ʹ��C6D6��Ϊ��Ӧ����ʱ����Ӧ���ɵı��������C6D5��N��SiMe3��2�к������Dԭ�ӣ������˱��Dz��ﱽ������Դ����������о������������ı���ͬϵ��ķ�Ӧ���ԡ���Ϊ����ʱ��Ŀ����ﱽ��������IJ���Ϊ68%���ױ�Ϊ����ʱ������Ϊ��λ����λ�����ܲ���Ϊ61%���ڶ��ױ�Ϊ����ʱ����Ӧ�����õ�12%�ļ�λ�������
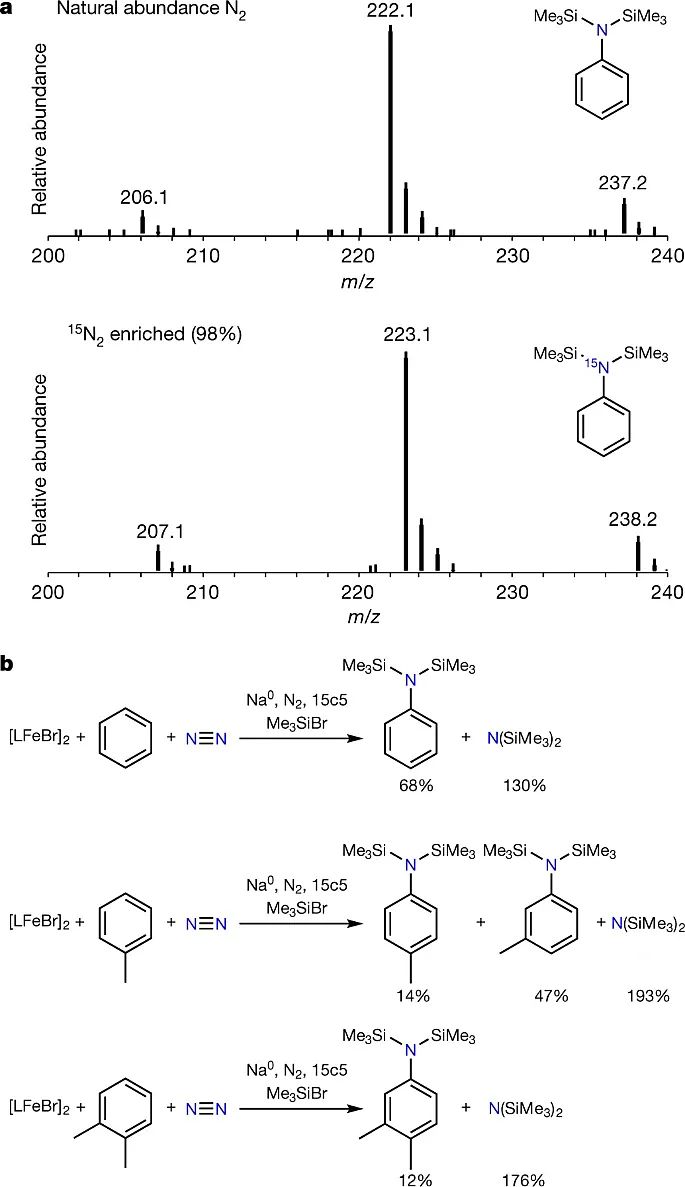
ͼ5. ͬλ�ر��ʵ�鼰�������ͼƬ��Դ��Nature
����ƪ�����У�Holland�����鱨����һ���������Ϊ���������͵�����Ϊ��Ӧԭ�����ϳɱ�����������·�Ӧ����Ȼ֮ǰ�������������������������������Ľ���ż����Ӧ�Լ��Ǿ���İ�����Ӧ���������û���ͪ�Ͱ����ϳɱ�����Nature, 2020, 584, 75�C81������Ķ���ϸ�����Ѿ������˱���������ĺϳɲ��裬������������ӷ�Ӧԭ�ϵĽǶ���˵�����ñ��͵������ϳɱ�����Ȼ�����������������ǵ���Ӧ���������õ����������÷�Ӧ��Ȼ���г�Ϊһ����Dream Reaction����DZ�ʡ����ǣ��÷�Ӧ������Ҫ�õ����Լ�ǿ�Ļ�ԭ�����Լ�ǿLewis�ᣬ����Ҫ�ϸ���ˮ�����Լ�����-100 ��ķ�Ӧ��������ʵ�ʲ����Ƕ��������÷����ڶ����ڻ������кϳ�Ӧ�ü�ֵ��������ˣ��÷�Ӧ���������������̵�������̼�����������ԳƵ���һ����̱�ʽ�Ĺ�����������һ��С������������д��ƪ����ʱ������������ͼ5b�������е������������һ�¡��������У�������ָͼ5bΪC6H6 ��C6D6��1:1���İ�����Ӧ��ͼ5cΪ�����ױ����ڶ��ױ��İ�����Ӧ����ʵ����ͼ5bȴ�DZ�ͬϵ��ķ�Ӧ����Ȼ������©����5b����5c��dz���5b���ڷ��ָô��������ͨѶ����Holland���ڷ����������ʼ������������Ӻ��߾��յ���Holland���ڵĻظ���������̾��Ч��֮�ߡ�
Coupling dinitrogen and hydrocarbons through aryl migrationSean F. McWilliams, Daniël L. J. Broere, Connor J. V. Halliday, Samuel M. Bhutto, Brandon Q. Mercado, Patrick L. HollandNature, 2020, 584, 221�C226, DOI��10.1038/s41586-020-2565-5
https://www.x-mol.com/university/faculty/1292