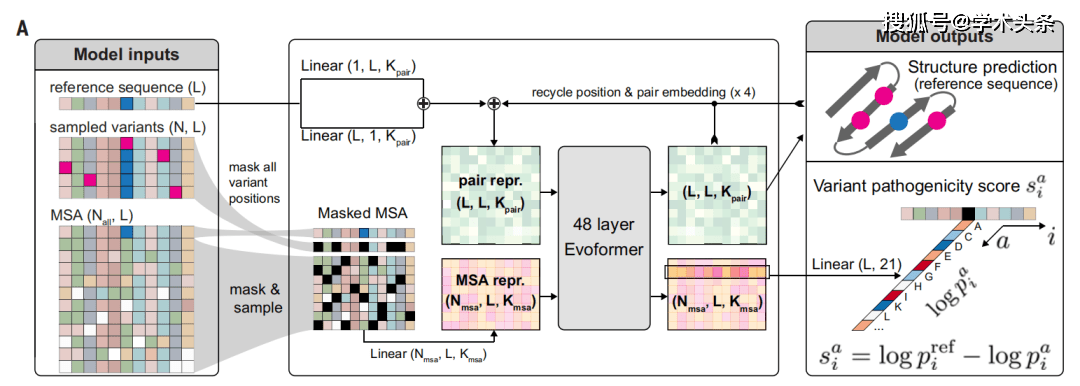
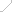具体来说,AlphaMissense 成功预测了 19233 个标准人类蛋白质的 216 百万种可能的单一氨基酸变化的致病性,得到了 7100 万个错义突变的预测。随后,AlphaMissense 更是成功预测出 89% 的错义突变,其中 57% 可能是良性的,32% 可能是致病的。
相关研究论文以“Accurate proteome-wide missense variant effect prediction with AlphaMissense”为题,已发表在权威科学期刊 Science 上。
在一篇同期发的观点文章中,爱丁堡大学的计算蛋白质生物学教授 Joseph A. Marsh 和剑桥大学研究院兼维康桑格研究所细胞遗传学负责人 Sarah A. Teichmann 评价道:
“虽然该研究无疑对变异解释和优先处理有所帮助,但重要的是不要将这些标签与这些术语具体的临床定义混淆,后者依赖于多条证据。”
值得一提的是,Google DeepMind 已经将 AlphaMissense 的所有预测免费提供给了研究社区,并开源了 AlphaMissense 模型的代码。
成功预测89%的错义突变
错义变异是指一种可以改变蛋白质氨基酸序列的遗传变异。致病性错义变异会严重破坏蛋白质功能,降低生物体适应性,而良性错义变异的影响有限。
在超过 400 万个观察到的错义变异中,仅有约 2% 被临床分类为致病性或良性,对剩余未知的变异进行分类是人类遗传学中的一个重要挑战。缺乏准确的错义变异功能预测限制了罕见疾病的诊断率以及针对潜在遗传原因的临床治疗的开发和应用。
虽然多重分析变异效应(MAVEs)系统地测量蛋白质变异的效应并可以准确预测变异的临床结果,但 MAVEs 实验需要高昂的费用和劳动力,因此蛋白质组范围内的变异致病性调查仍然不完整。
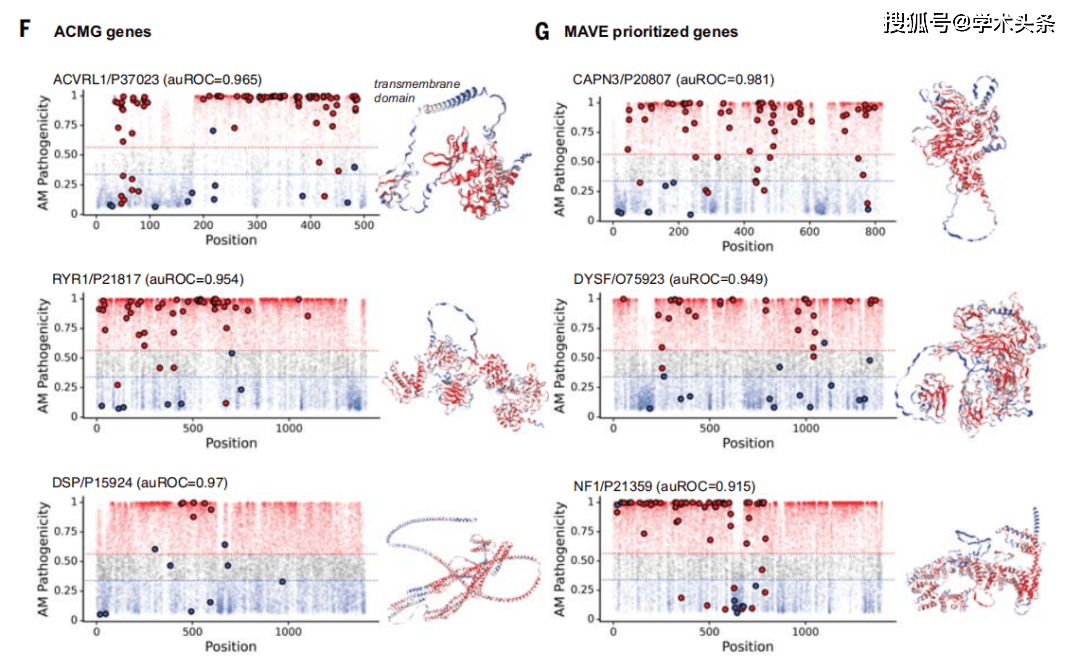
机器学习方法可以通过利用生物数据中的模式来预测未注释变异的致病性,从而缩小这种变异解释差距。AlphaFold 的成功已经证明,可以使用蛋白质序列作为输入来预测大规模的高精度蛋白质结构,而这种蛋白质结构模型可以作为理解蛋白质生物学其他方面(如变异致病性)的基础。
在该研究中,借助 AlphaFold 的方法论,AlphaMissense 结合了现有策略的三个元素:
1)基于人口频率数据的弱标签训练,避免使用人工注释,从而避免了循环性;
2)通过使用无监督的蛋白质语言建模任务来学习在序列上下文中条件化的氨基酸分布;
3)通过使用 AlphaFold 派生的系统来整合上下文。
据论文描述,AlphaMissense 的训练分为两个阶段:结构预训练和变异微调。其中,预训练阶段与 AlphaFold 中描述的相同,但在蒙版多序列比对重建损失上增加了更高的权重;在微调期间,模型被优化,可以同时预测变异的致病性和参考序列的结构。
以往研究表明,良性训练变异是基于在人类和其他灵长类物种中频繁观察到的变异,这些变异是根据 PrimateAI 方法来定义的,而致病性训练变异则是从未在人类群体中观察到的变异中进行抽样,抽样权重取决于三核苷酸上下文和基因。
AlphaMissense 不预测突变对蛋白质结构的影响或对蛋白质稳定性的其他影响。相反,它利用相关蛋白质序列的数据库和变异的结构背景来生成一个介于 0 和 1 之间的分数,这个分数评估了变异可能是致病性的概率。连续的分数允许用户选择一个阈值,以符合其精确度要求,从而可以将变异分类为致病性或良性。
AlphaMissense 将 7100 万个可能的错义变异中的 89% 分为可能致病或可能良性两类。相比之下,仅有 0.1% 的变异已被人类专家确认。
AlphaMissense 在广泛的遗传和实验性基准测试中实现了最先进的预测,而且完全没有明确地在此类数据上进行训练。
此外,该模型在用于分类 ClinVar(一个关于人类变异与疾病关系的公共数据存档)中的变异时表现也优于其他计算方法。
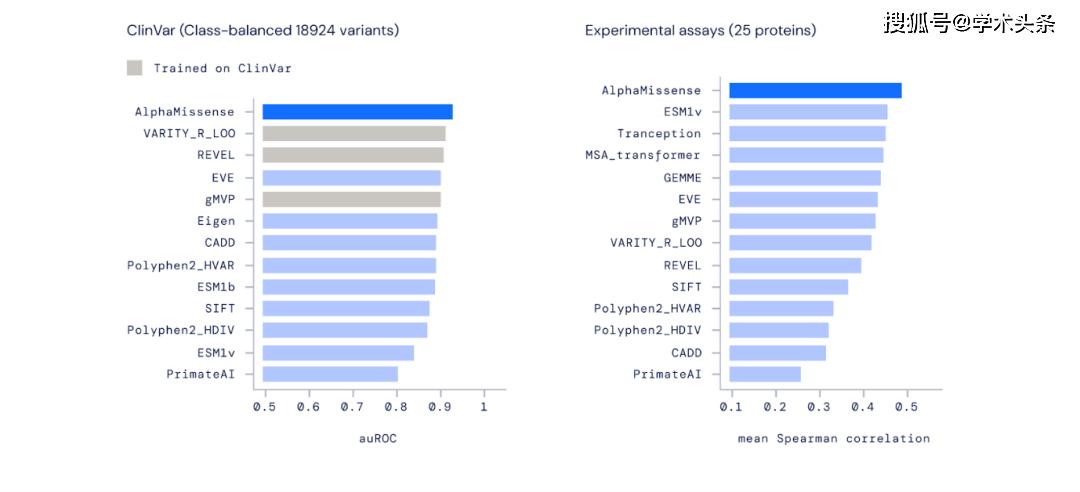
有望解决人类遗传学难题
毫无疑问,AlphaMissense 的预测阐明了变异对蛋白质功能的分子影响,这有助于识别致病性错义突变和未知致病基因,同时提高罕见遗传疾病的诊断率。此外,AlphaMissense 还将促进专门的蛋白质变异效应预测器的进一步发展。
然而,Marsh 和 Teichmann 也指出了 AlphaMissense 的一个局限性:目前其预测器的结构组成部分并没有考虑到大多数蛋白质会组装成具有多样四聚体结构的复合物或凝聚体。对于形成复合物的蛋白质突变,可能会导致疾病,但仅考虑单体结构时这种方式可能并不明显。
此外,尽管许多与疾病相关的突变通过蛋白质不稳定性或复合物组装的破坏导致功能丧失,但在其他情况下,突变蛋白质通过显性负效或增效效应引发疾病。
因此,有趣的是观察 AlphaMissense 在非丧失功能变异方面的表现,这些变异通常对氨基酸的干扰较小,几乎所有先前测试过的变异效应预测器(VEPs)都难以准确预测这类变异。
最终,结合蛋白质四聚体结构的信息,可能可以通过预测蛋白质复合物结构的算法来实现,这有望在变异效应预测领域带来更大的改进。