ת¼���Ŵ���Ϣ��DNA���ݸ�RNA�Ĺ��̣������ķ���ĵ�һ����ת¼������ѧ��չʷ��һֱ�����ž������ص����ã�Ҳ��ĿǰӦ�����ĸ�ͨ�������������֮һ��
��Ϊת¼��ѧ�о��Ļ��������ڱ������ķ�������������ѧ���̵Ĺؼ���Ҳ����Ϊ�����һ�ַ����ֶΡ������ר���У����ǽ�����̽��ת¼��ѧ�����ֻ��ڱ������ķ����������ֱ��Dz�����������Ʒ�����WGCNA�������������ǵ���ƪ���½��۽���ת¼��IJ����������һ���䷽��ͨ�������Ƚ�����֮��Ļ������ˮƽ����ʶ��ؼ�������ѧ�仯������������С��Χ��Ѱ��Ŀ�����ͨ·��
��������
��ת¼�����ݷ����У��������������ķ�����������������ĺ�������������ͬ��𣨴����飩֮��ı�������Ƿ������������ڣ�������IJ��졣
ͨ�������������ı仯������Fold change���Ͳ������ֵ��P/FDRֵ��������ȷ����Щ������������������ֳ������IJ��죬������Щ���������ĸ��������ȷ������ϣ���̽����ص�����ѧ���̺ͷ��ӻ��ơ�
ת¼����������Ҫ����
���������ͳ�ƣ�
����ÿ�������ת¼���ı�������ͨ������ԭʼ reads count ��FPKM��ÿǧ���ÿ������Σ���TPM��ÿ����ת¼�������ȷ�������չʾ��
ԭʼ reads count ��ʾ��ת¼��������reads��Ŀ�����ܲ������ͻ��ȵ�Ӱ�졣Ϊ��֤��������ȷ�ԣ������ȶԲ�����Ƚ���У�����ٶԻ����ת¼���ij��Ƚ���У������ñ������FPKMֵ��TPMֵ���ٽ��к���������
����������ɸѡ��
ʹ��ͳ�Ʒ�������DESeq2��edgeR�ȣ��Ƚϲ�ͬ������Ļ������ˮƽ��ɸѡ���������Ļ����õ�ɸѡָ��������챶����FC���Ͳ������ֵpֵ/FDR��
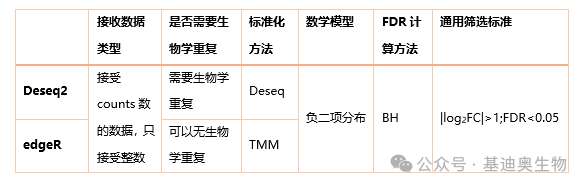
FC��Fold Change����
FC��ʾ������Ʒ��ͬһ���������ˮƽ�ı仯����������ӳ��ʵ����Ͷ�����֮��ı��������졣FCֵԽ��ʾ�������ı仯Խ�������ڲ����������У�ͨ�����趨һ��log2FC��ֵ���������1��2����ɸѡ����������
pֵ��
pֵ��ͳ��ѧ��������ʾ�۲쵽�������������������ݲ����Ƿ�������һ��ָ�ꡣpֵԽС����ʾ����ı������Խ������Ȼ����pֵ���������ܵ����ؼ�������Ӱ�죬�����ʵ��Ӧ������Ҫ�������ָ������ۺ��жϡ�
FDR��False Discovery Rate����
FDR���ڿ��Ƽ����Խ���ı������������б�������Ϊ����������Ļ����У������в���Ļ�����ռ�ı�������ת¼������У�������Ҫ�Դ���������м�����飬���������FDRУ������ʹ���μ���ļ������ʺܵͣ��ۻ��ļ�������Ҳ���ܸܺߡ�FDR�ļ���ͨ������Benjamini-Hochberg������ͨ����ÿ�������pֵ���������У�����Ӷ��������յļ������ʡ�
һ�������ɸѡ FDR<0.05 �� |log2FC|>1 �Ļ���Ϊ�����������Ҳ���Ը��ݾ�������ʵ��ſ�������ɸѡ����
���ò���������ӻ�ͼ��
�������ϵ�һϵ�в�������������ɸѡ����һ��������������������Ļ�����Щ��������������Ǿ�ģ����������Ҫ����ֱ�۵�ͼ�ν����ݿ��ӻ����Ա���õز�������ѧ���⡣
���õ�ͼ������״ͼ����ɽͼ����ͼ��Τ��ͼ�ȣ����Ӹ߶�һ���ͼ����С����ͼ���������ɢ��ͼ���״�ͼ�ȵȣ���������չʾ��ϸ�ڻ���ӷḻ���������г����������϶���ͼ����ϵķ�ʽ����Ƕ�չʾ������������
01
��״ͼ
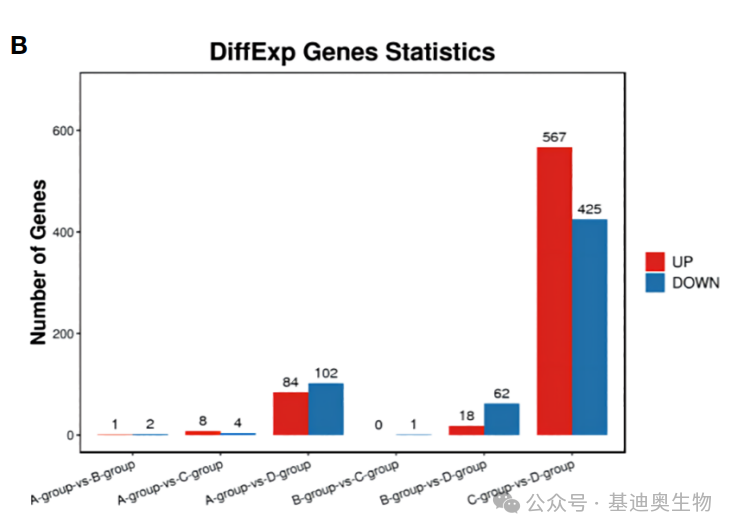
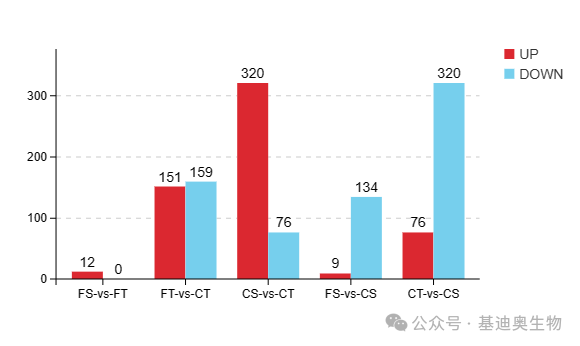
��״ͼ��Ҳ������ͼ����ת¼�������������������Ҳ�Ǹ�Ƶ���ֵľ���ͼ�Ρ��������ڱȽ����������������ݣ��粻ͬʱ�����������ͨ��ֻ��һ��������������С���ݼ�������
ͨ����״ͼ�����ǿ���ֱ�۵ؿ�����������ϵ����µ��IJ�������������Ӷ���ӳ��������̵Ĵ������ƣ�Ҳ������������֤һЩԤ�ڵ�ʵ������
02
��ɽͼ�Ͷ����ɽͼ
��ɽͼ��һ������չʾ�Ƚ������������������ͼ�Ρ���ͼ�α�����ɢ��ͼ��һ�֣����ܽ�ͳ����������ֵ��-log10��FDR�����Ͳ��챶����log2FC�����ϣ��Ӷ��ܹ���������ʶ����Щ�仯���Ƚϴ��Ҿ���ͳ��ѧ����Ļ���
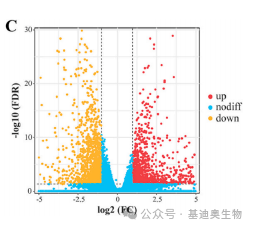
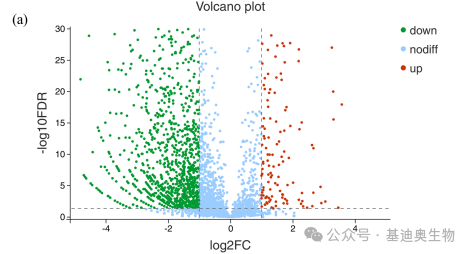
��Ȼ��ɽͼ��չʾ��ϸ�ںܷḻ�������ľ���������ֻ�ܱȽ�����������ݡ�������Ҫ���ܵ����ͼ���ֲ������ȱ�ݡ�
�������ɢ��ͼ��Ҳ�ж����ɽͼ������һ����չʾ����Ƚ���IJ������ֲ���������ĺ�����Ϊ�Ƚ�������ƣ�������Ϊlog2FC������ʱ���Զ�ɸѡPvalue/FDR<0.05�IJ��������Ļ������չʾ��
03
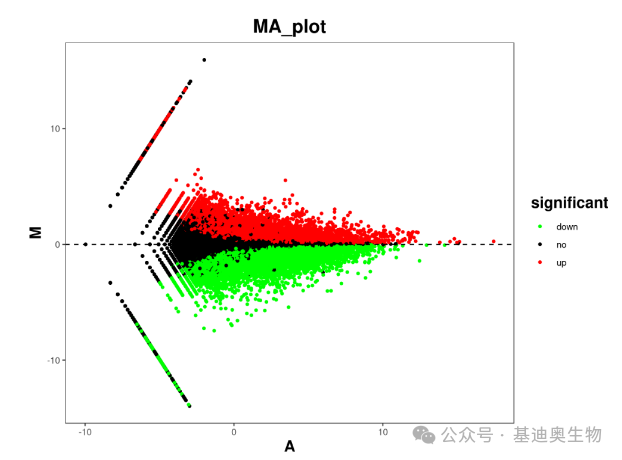
MAͼ
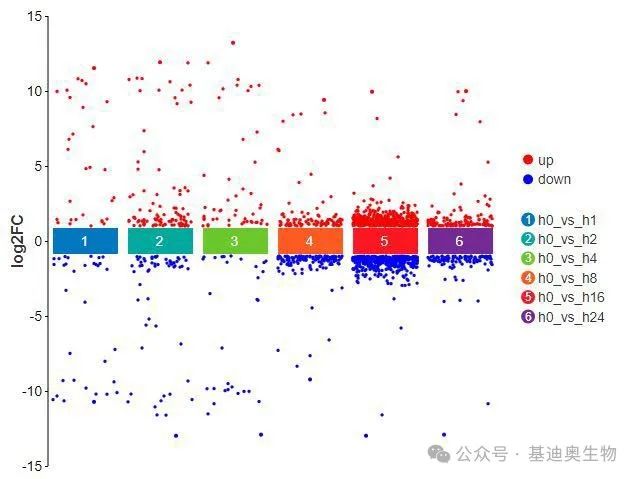
MAͼ��Minus-versus-Add������һ������չʾ���������ɢ��ͼ��������microarray��оƬת¼��������ݷ����лᱻʹ�á�
��ʱ���о���Ա��Ҫ���õ�ͼ�Ƚ�����������IJ��죬X����������ֵ�ľ�ֵ������a+b��2��Y��������IJ�ֵ����b-a��һ����add��һ����minus�������MA��Minus-versus-Add��plot�ˡ�
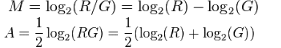
����������ʽ��M�ͱ����log2��FC����A������������ƽ��ǿ�ȣ�������������һ��Ϊ���챶��Ϊ2�����ϣ���pֵС��0.05���ĵ㲻ͬ����ɫ����ʾ������Ʒ���������ԡ�
��һ���㣨��һ������Yֵ��0��˵�����������û�в�𣬵���X����ֵԽ��˵����������ľ�ֵԽ����ô����һ�����X����ֵ�ܴ�Y�����ֵҲ�ܴ��ʱ��˵����������ƽ���������ߣ�����ܴ�Ļ�����ζ���������һ��������һ�����ž��˵ı��������������������Y�����ֵ�ܴ���X����ֵ��С��˵�������п�����С������Ļ������ı仯�����ϴ�ı���������
04
������ͼ
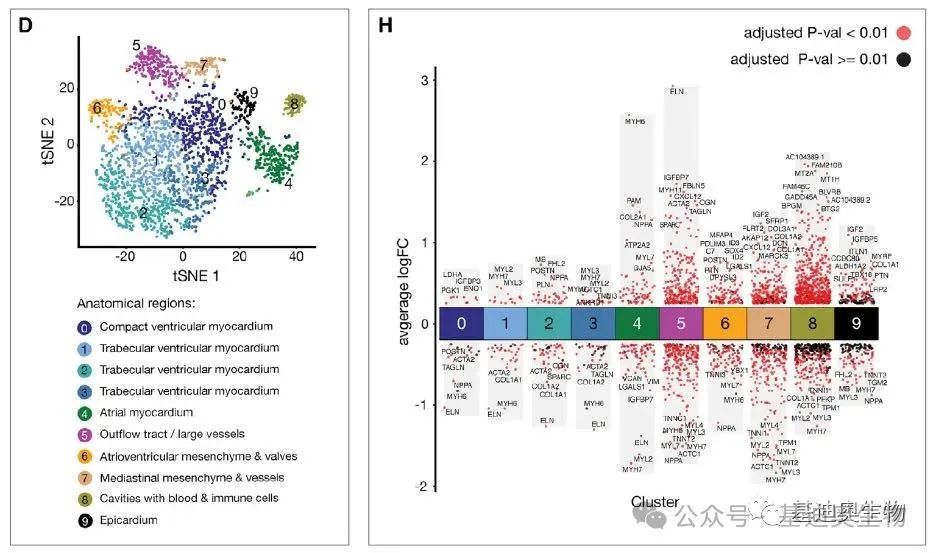
�ڲ�����������ɺ����ǿ��Խ�����Ȥ�ػ����þ�����ͼ��չʾ��ÿ��С�����ʾÿ�������ڲ�ͬ�����еı����������������Խ����ɫԽ��Ϸ�����������ͼ��ʾ�Բ�ͬ��Ʒ�ͻ���ľ�����������
��ͼ����ֱ�۳��ֶ�������������ȫ�ֱ������仯����չʾ����ͬ�����Ļ���������ơ�����֮�⣬���ǻ����Ը��ݾ���Ľ�����۲��������ظ��ԣ����Ƿ��в�ͬ������������ൽһ�𣬻����Դӻ������ĽǶȣ�����Щ������бȽ�һ�µı���仯�������ܹ��ڹ���Ԥ���ϸ�����һЩ������
�ڻ��ƾ�����ͼʱ�����Dz��ɱ����Ҫ˼��һ�����⣬���ǹ�һ�������⣬��һ������˵���ǽ�һ������ͨ����һ��������ʹ����Ͼ�ֵΪ0������Ϊ1�ı���̬�ֲ�����ô������ѡ���һ������ʽ�أ�
a�����о�һ����
��ÿһ����ֵ�ֱ�������ʹ����ϱ���̬�ֲ���ͨ���������Ի���Ϊ��λ���۲���Щ��������ֵ�ı仯�����磬A�����������10�仯��20��B�����������100�仯��200�����Ǹ��������DZ仯�ı�������ʱ����Ӧ��ѡ���Ի���Ϊ��λ���й�һ����
��ˣ��������һ���������������̶ȵس���ÿ������ı仯��Ϣ������һ�����߱���Ļ����ڸ�����������ı仯���ڻ�����ͼʱ��������õ�һ�ֹ�һ�����ԡ�
b�����о�һ����
��ÿһ����ֵ�ֱ�������ʹ����ϱ���̬�ֲ������������ͨ������ı��������۲��������ظ��Ժû����������࣬��ʱ����ͨ�����й�һ��������
c���к��ж���һ����
�����е���������һ������ʹ����ϱ���̬�ֲ���������ø߱���Ļ���������ķ�����������ã���ʱ����ѡ���������ֵ��һ����
d��������һ������:
��ʾֻ��ͨ����ɫ�Ľ������۲����ı������仯������ѡ������һ��������
05
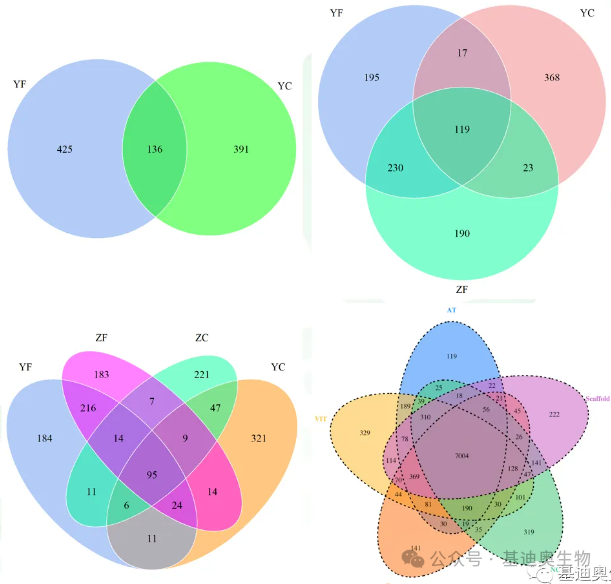
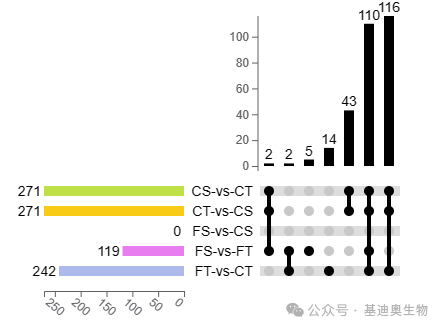
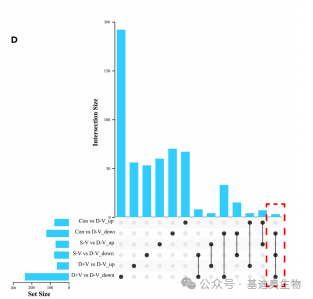
Τ��ͼ��Upsetͼ
ά��ͼ��upsetͼ�Ƕ�����������֮��Ƚϵõ��IJ��������еļ��ϣ��ص����ּ��Dz�ͬ�����¸��������б���ͬ���صĻ��������IJ�������ij�ִ������ض����صĻ�������ά��ͼ��upsetͼ���ǿ��Էֱ�Թ��л����л���������ھ�
Τ��ͼ���Ŵ���Ѿ���İ���ˣ�Ҳ�ȽϺ����⡣����Τ��ͼһ��ֻ�ܻ���2-5���Ƚ�����չʾ������ıȽ���������Τ��ͼ�ͱȽϷ��Ӳ������ˡ���Upsetͼ���������Τ��ͼ�������Ը��õ�չʾ6�������ϵıȽ���Ľ����������
06
�״�ͼ
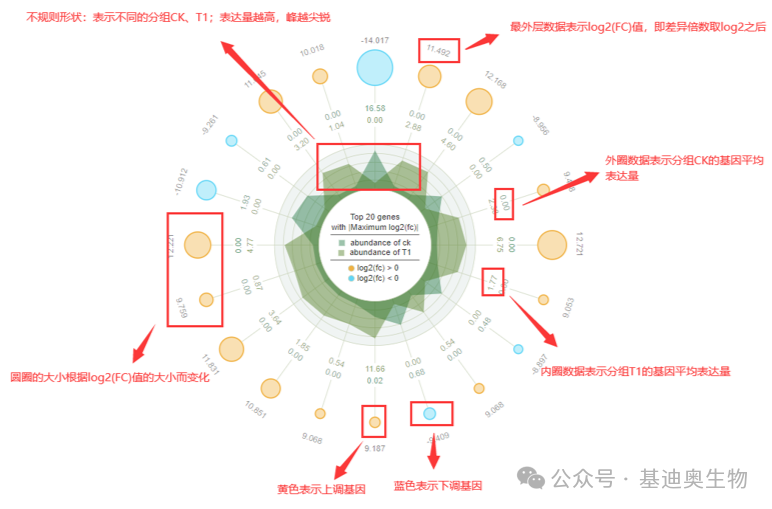
�״�ͼ���Դ�ͬһ�㿪ʼ�����ϱ�ʾ���������������������Ķ�άͼ������ʽ��ʾ��������ݵ�ͼ�η�����ͼ�Ͽ���һ����չʾ��Ϣ������log2��FC��ֵ��ƽ�������������µ�����ȡ����ǿ���չʾ�������������Pֵ����Qֵ����չʾ����̶�����TOP n�������Ϣ��
ת¼��������Ӧ��
������ô�����ۣ����������Ǿ�Ҫ��ʼ��ʵս���ˣ�Ҳ����ѧϰ��ν�����������õ������Լ����о��С��ڿ�����ƪ���º��㶨���Ȼ���ʣ���ʶ�����ƼIJ������Ҳ�ܱ�Ӧ�õ�������

��ƪ���Խ��տƼ���ѧ�Ļ��ϰ¿ͻ������£���2024��2�·�����Plant Physiology and Biochemistry��IF��6.5����־�ϡ�����ͨ��RNA��������ɣ����Morus alba��ҶƬ�ڲ�ͬŨ�����������µ�ת¼������˲��������ּ�ڽ�ʾɣ��Ӧ����в�ȵķ��ӻ��ơ�ʵ��һ��������5������������ȱ��T1�����ж�ȱ��T2����������CK�������ж���T3�����������ж���T4��������CKΪ���ա�
�����ڵ�һ��������չʾ��ת¼�����ݵ������Լ�������������Ϣ������reads������������ɷַ�����PCA���������������ͼ��FPKM �ܶ�ͼ������С���ٱ���ͼ����������Τ��ͼ��
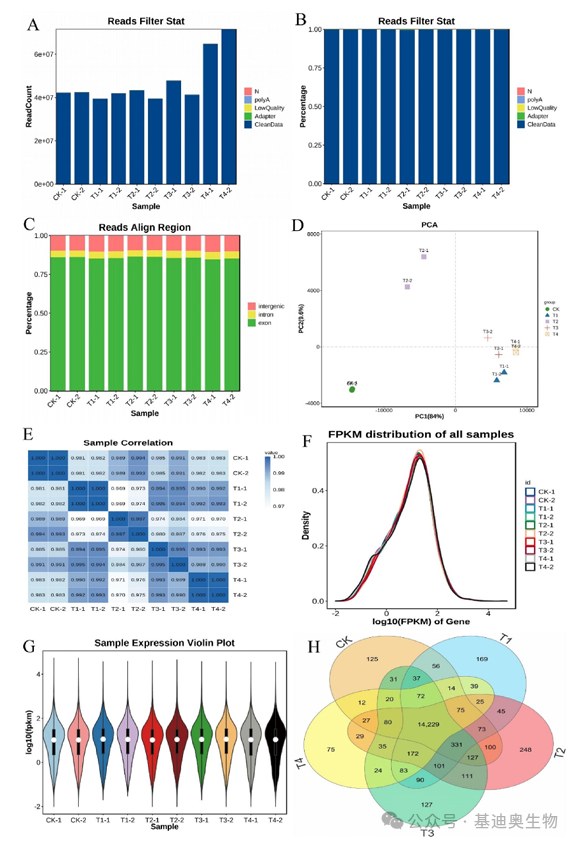
ͼ1 ת¼�����ݵ�����ͳ��
�ڵڶ����֣����߿�ʼ��ת¼�����ݽ��������ϵIJ����������4��ʵ������CK�������������������ɸѡFDR<0.05��|log2FC|��1�Ļ���Ϊ����������DEGs����ͨ����״ͼ��Τ��ͼ�Լ���ɽͼ���⼸ǧ������������չʾ��
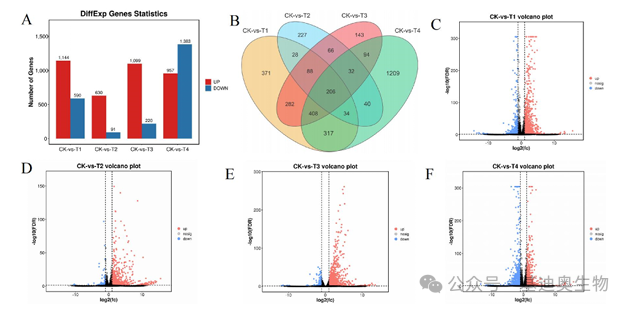
ͼ2 ��ͬ��ˮƽ��ɣ�������µ�����������DEGs��
�����ţ����߶�ɸѡ������DEGs������GO��KEGG�����������ֱ�����pֵ�������ӣ�enrichment factor����������ͼ��pֵ�����˸����������̶ȣ�p<0.05����ɫ�������������������������Ӵ����˸�term/pathway��Ӧ�IJ�����������Ŀ������鱳���и�������һterm/pathway����Ŀ�ı�ֵ����ֵԽ�߸����̶�Խ������


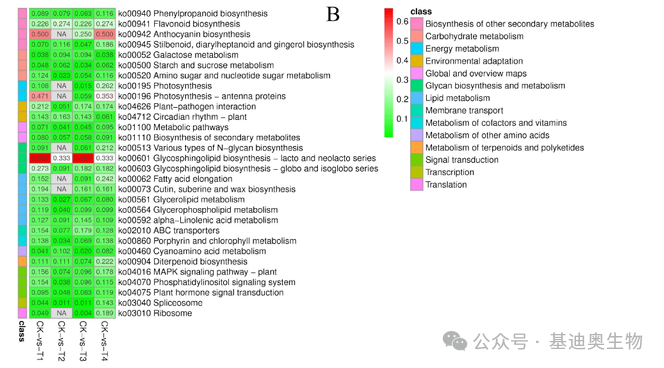
ͼ3 DEGs��GO��KEGG������ͼ
�ڵ������֣����߿�ʼ�۽�����Щ��ֲ�����в������������Ӧ�Ĺ��ܻ�����ת¼���Ӽ��塢ת�˵����塢���������ػ���ֲ�D�غ��źŴ�����صIJ�����������ϸ���ڽṹ��صIJ���������Ϳ�������ػ���ȡ�
����ɸѡ������ÿһ��������߶�������״ͼ�������ͼͳһ��ֱ�۵�չʾ�������ڲ�ͬ���������µı���ģʽ��ʹ�ò��챶����log2FC�����Ƶ���״ͼ����ֱ�۵ع۲�ÿ�ֻ���IJ������µ��������ʹ�þ�����ͼ����Է���չʾ�����ڲ�ͬ�����µı���ģʽ���졣
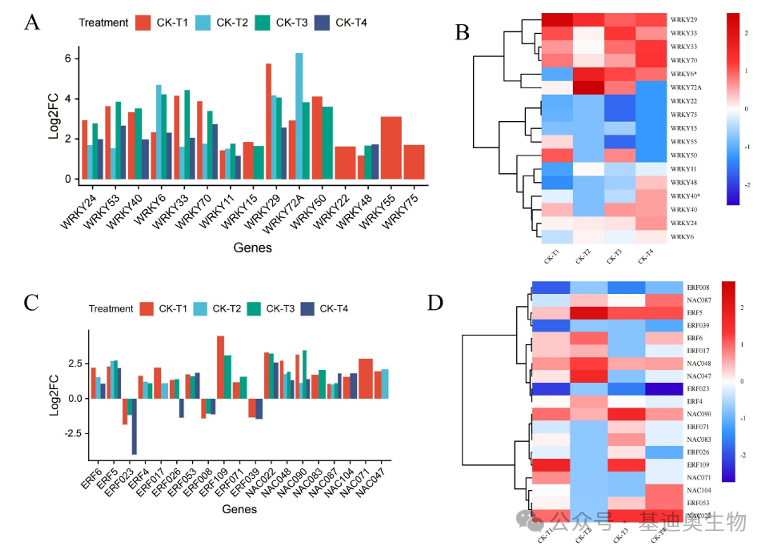
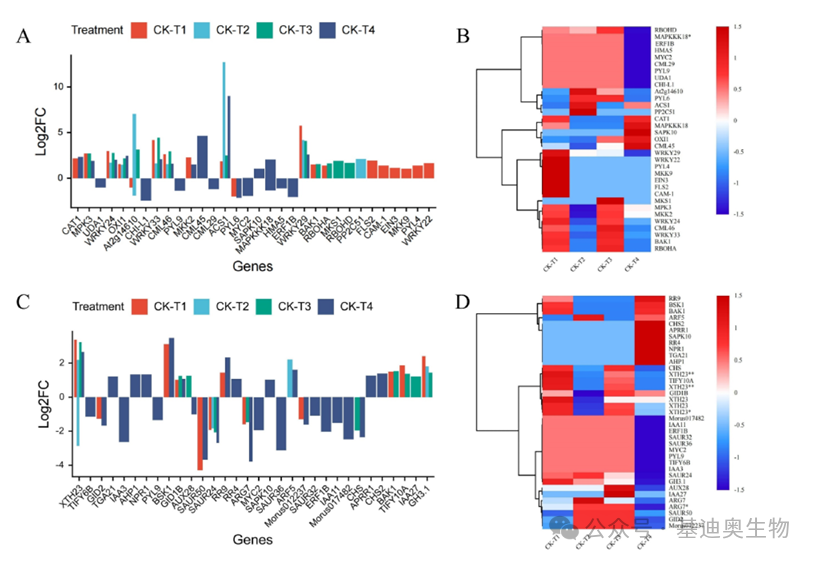
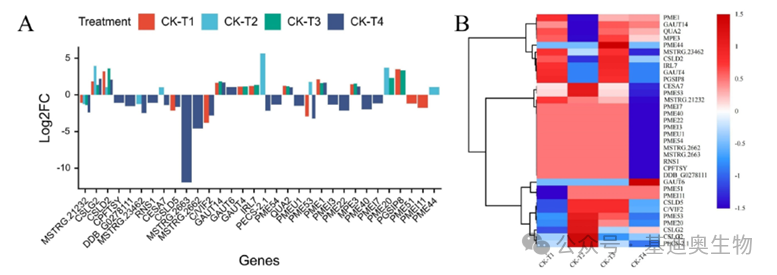
ͼ4 ���ֹ��ܻ���Բ�ͬ��������Ӧչʾ
�ڵ��IJ��֣���������ɢ��ͼչʾ��16���������RT-qPCR��֤�Ľ���������ʾRNA-seq��RT-qPCR����֮���������������ԣ�R2=0.88����֤����RNA-Seq����ĸ߶ȿɿ��ԡ�
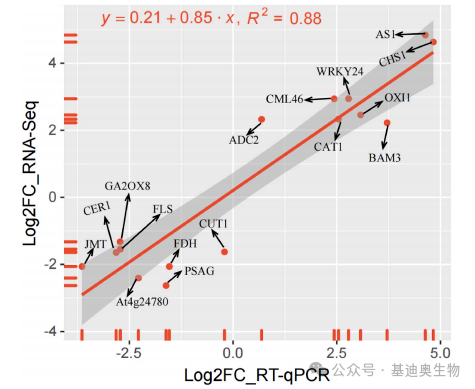
ͼ5 ����qRT-PCR������ɣ��16����������RNA-seq���ݽ��лع������֤��
������߽��֮ǰ�Բ�ͬ�����µ����������Լ���л����о����ܽ�������½��۲����Ƴ���һЩ������Ĺؼ���л;���������������Ļ�������ͼ��
С�
����һƪ����IJ������+������������ɻ�����������������£���Щ����������ͼ�ε����ã�Ϊ�����ṩ��һ���dz�ȫ��Ļ������������������ͬʱҲ�����Ǵ�����������ʾ��
�� ���ֲ���������ʹ�ã�ͨ����϶��ֲ������������ͼ�Σ�������ɸѡ��ֵ�����Ը�ȫ��ؽ�ʾ�������IJ���ģʽ��
�� �ص��ע��������ͨ·������֮ǰ�ı���ʵ���Լ������У�ѡ���ص��ע�Ļ�������ͨ·���з��������Ը�ȷ�ؽ�ʾ���ӻ��ơ�
�� ϵͳ�Եķ���˼·������ϵͳ�Եķ���˼·���ӻ��������쵽ת¼���ӡ��ź�ͨ·�ȶ������������������
������һ�������֪ʶ�����ǶԲ���R���Ե�����С����˵��һ���ֻ����������ء�û��ϵ����¼���ǵ�OmicsShare��ʹ������IJ������С���ߣ�������Ҳ���Էַ��Ӹ㶨��������������������ȡ�̳̰ɣ�
���������ɲ�������������qq.com��
�������ӣ�
https://www.omicshare.com/tools/Home/Soft/diffanalysis
����������ǻ��ϰ���ת¼����Ŀ��С��飬���ǵĽ��ⱨ���������������������������Լ������ݽ����ھ�������Omicsmartƽ̨�ϣ������Լ��������ݣ�ֱ�����ɸ��Ի����������������ӻ�ͼ�Σ��������ɸѡ����������˵�dz����з����ˣ�

��һ�����ǽ��ص����ת¼����ڱ������ķ����������������Ʒ��������ַ�����ʽ��ͬ�ڳ����IJ�����������ӡ��ߴ��ϡ������ڴ�����Ũ���ݶȻ���ʱ���ݶȵ���Ʒ���������������������½�����������Ҽǵù�ע���ǵĹ��ںţ�ǧ��Ҫ����Ŷ~