 |
|
 |
|
||||||||||||||||||



 ����������������NDA���ܻᱻ�ܾ���
����������������NDA���ܻᱻ�ܾ���





































































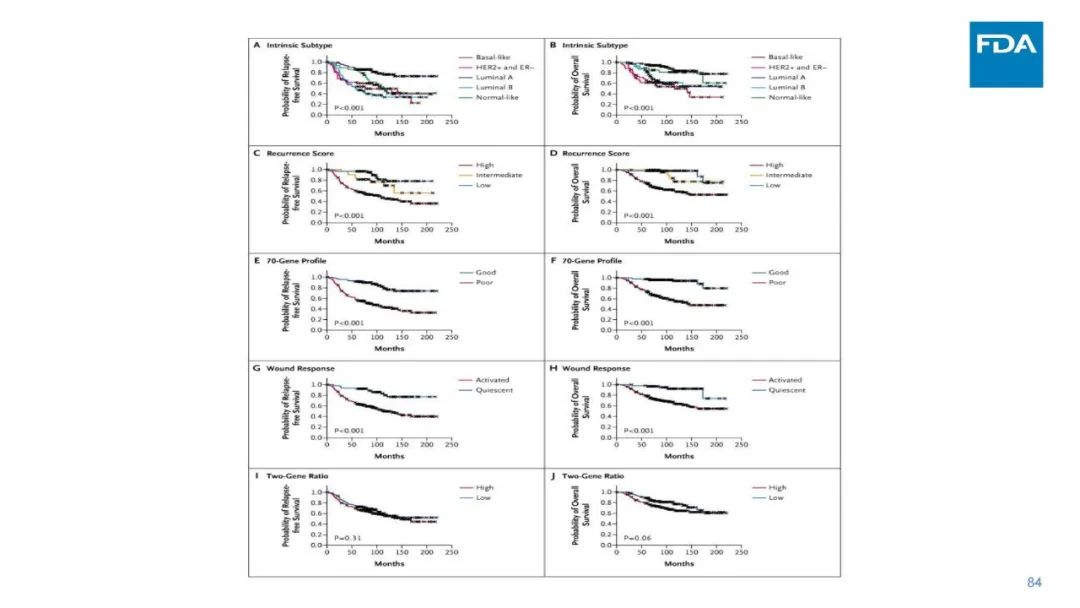





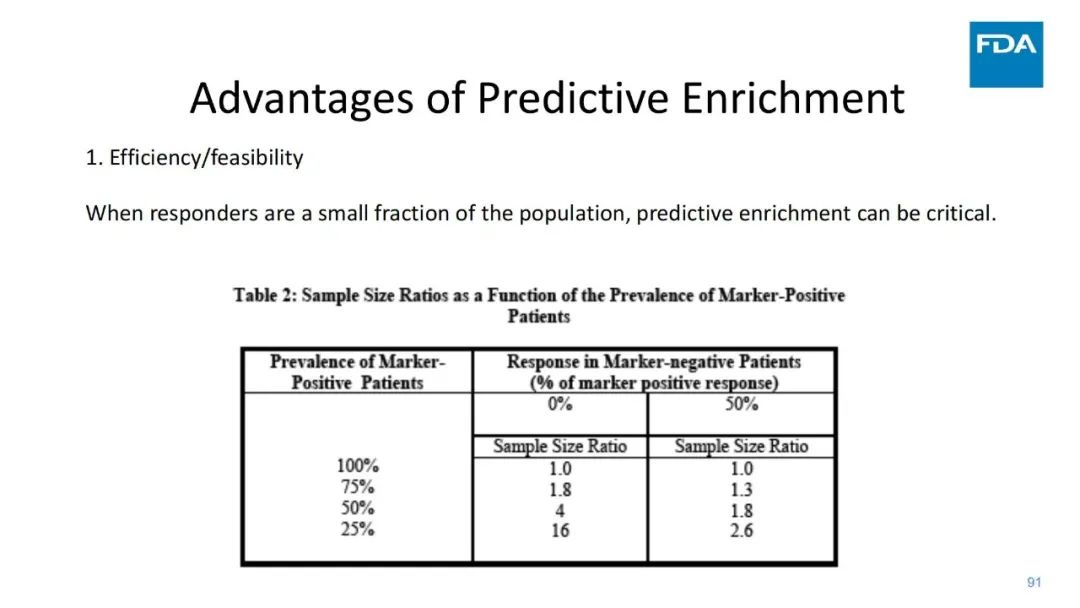




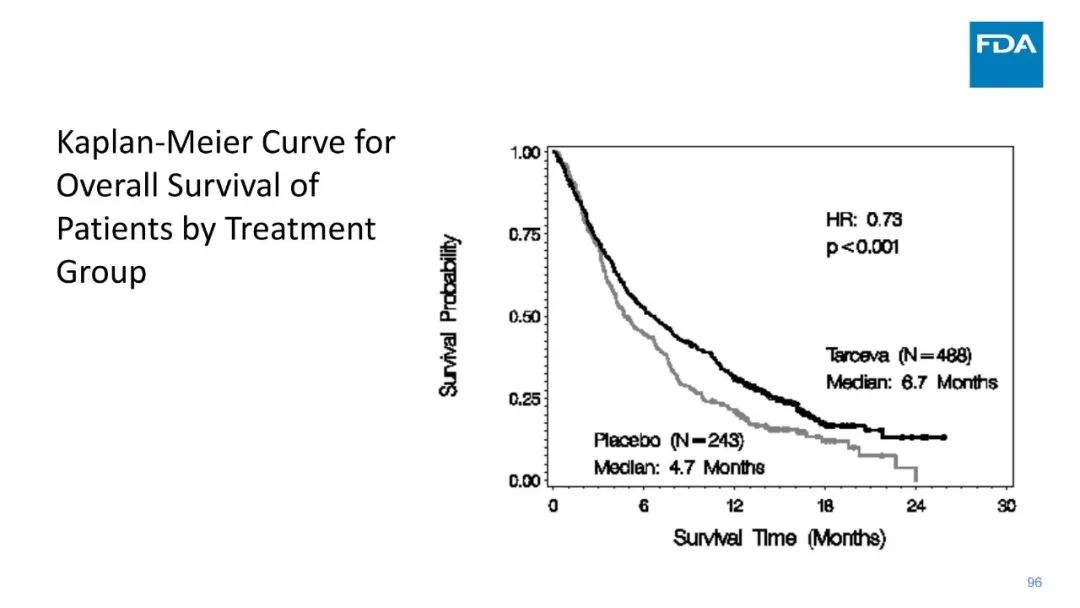
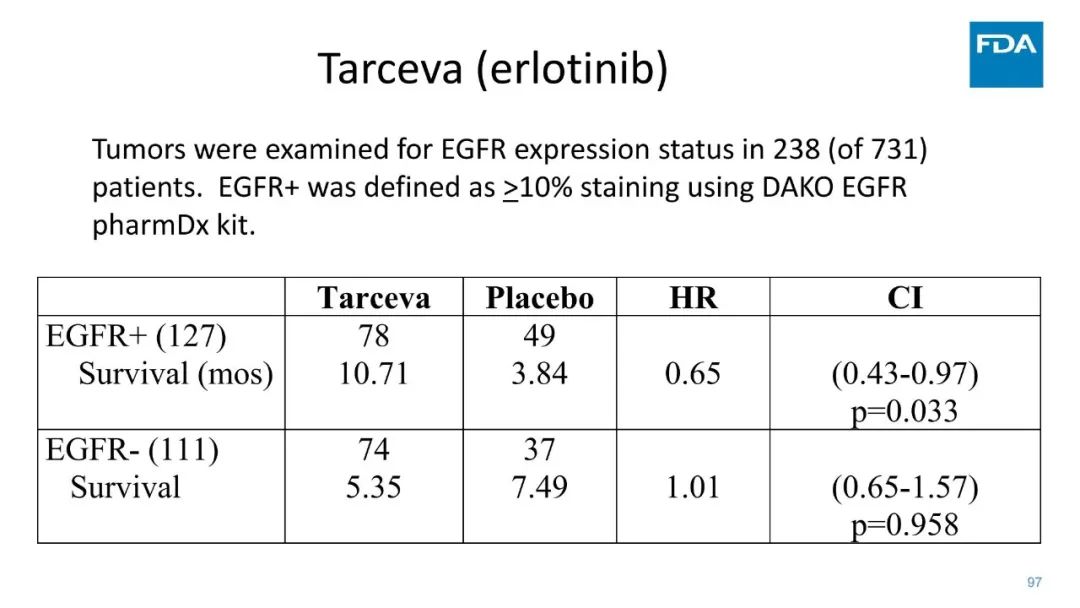
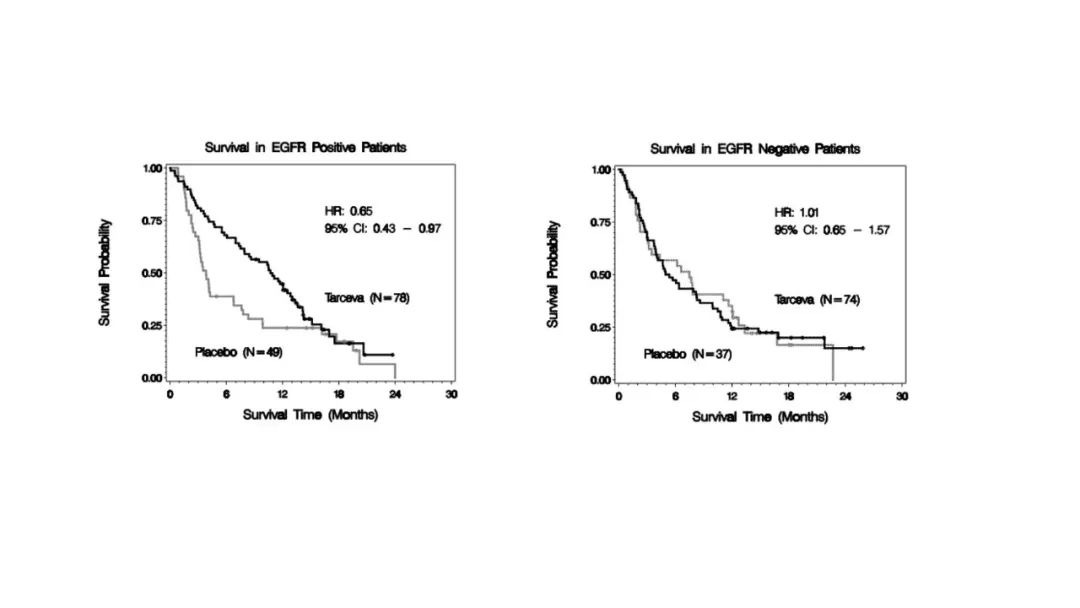



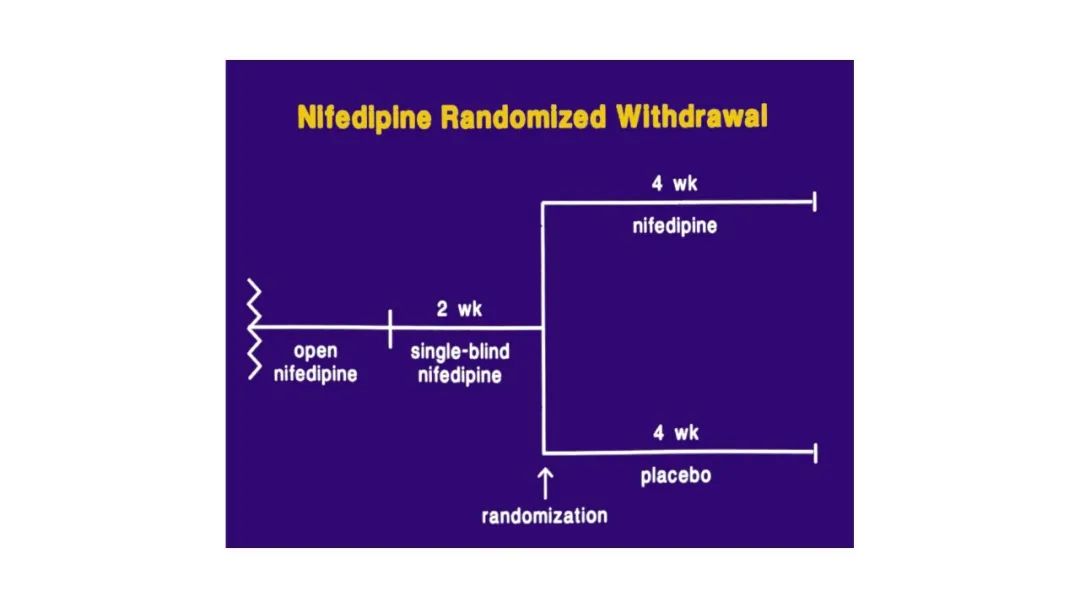






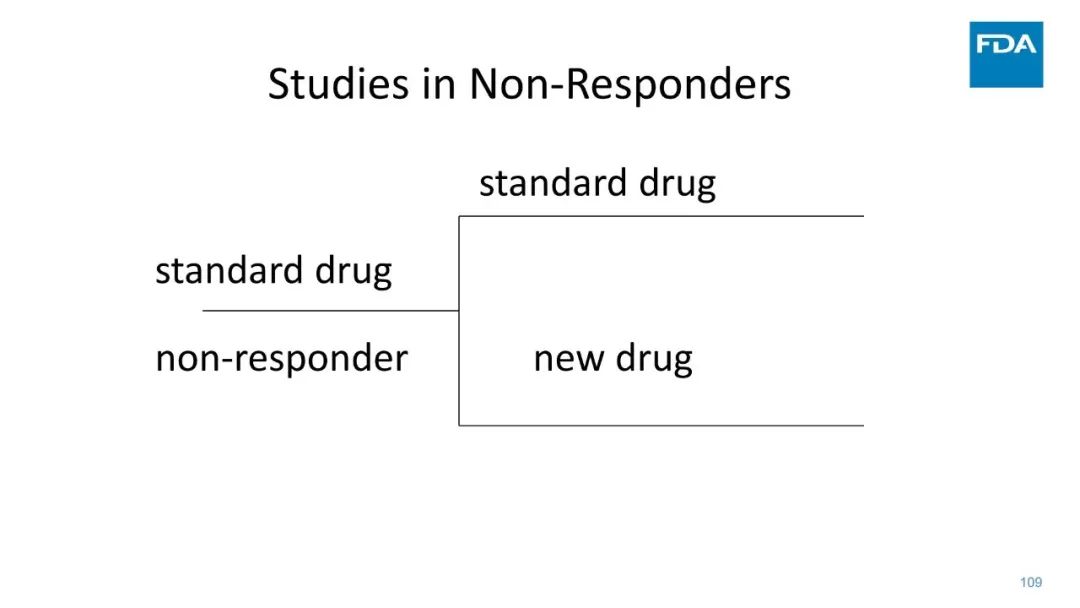

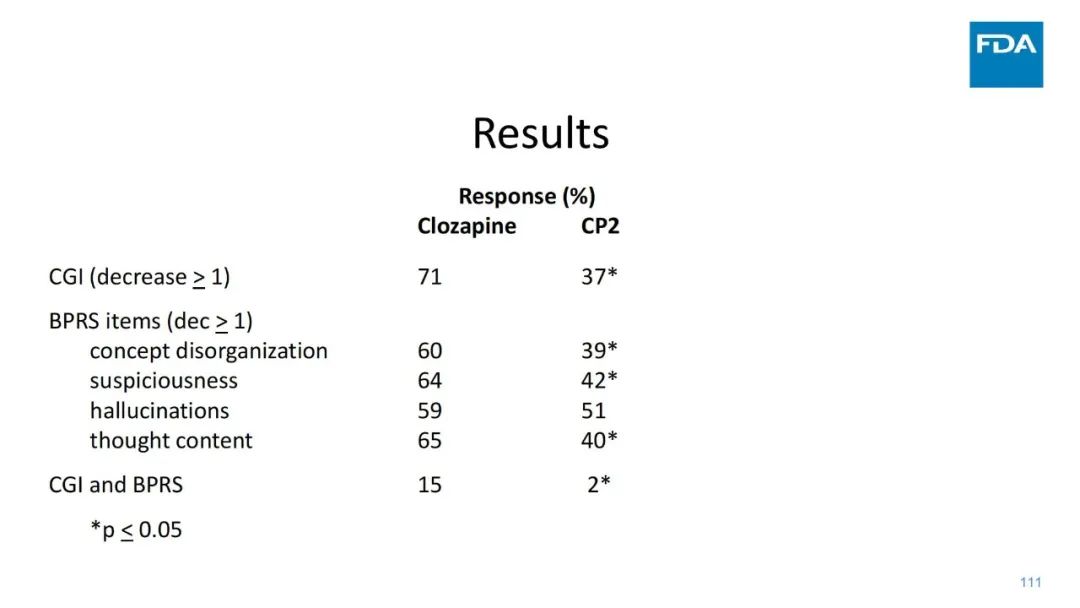

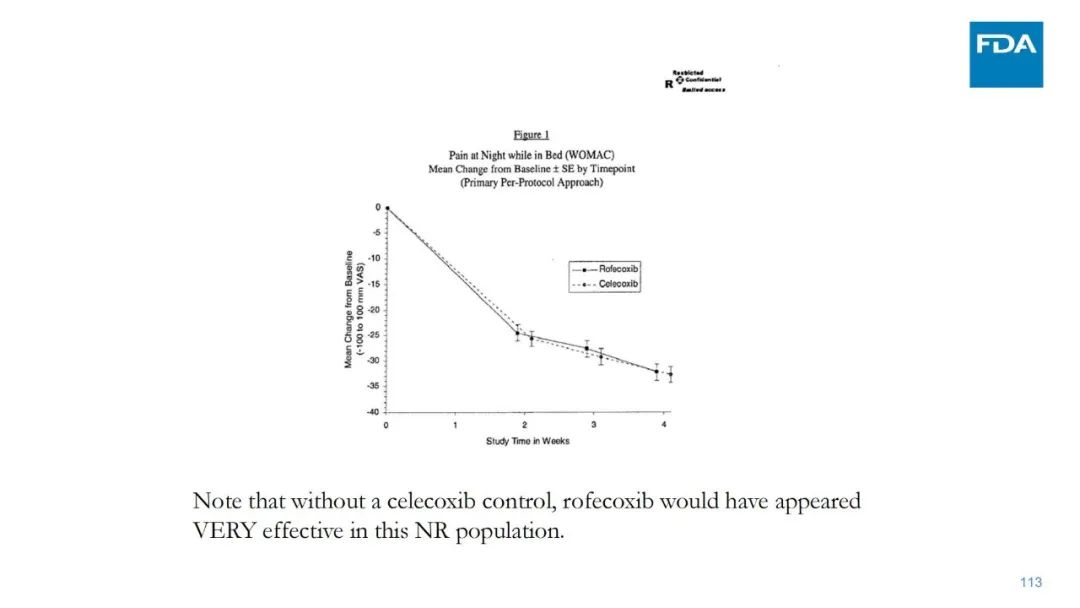
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||

|
|||||||||||||||||||||||||

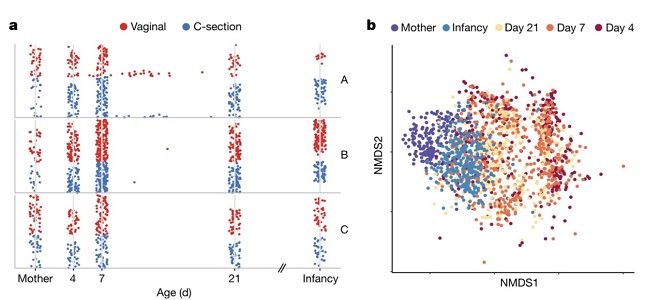






|
||||||||||
 |
|
 |
|
|
|
|
|
|
|
 |
 |
 |
| ��������������ϵ�����������������̨��������Ϊ��ҳ�������ղ��� |
| �����������ר������Ȩ���У�л������������ѡ�ǻ������������������ר������ϵ�� Copyright © 2026 ScholarsUpdate.com. All Rights Reserved. |
|
|
|
|||
| |
| ��Ŀ���� �� �����������ר�������Ƽ�����������ҽѧ |
|
|
|
|
|
||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||
|
||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
| ��������������ϵ�����������������̨��������Ϊ��ҳ�������ղ��� |
| �����������ר������Ȩ���У�л������������ѡ�ǻ������������������ר������ϵ�� Copyright © 2026 ScholarsUpdate.com. All Rights Reserved. |