ШЋЮФЫйРР
БОЮФзлЪіСЫУмЖШЗККЏРэТлЃЈDensity Functional Theory, DFTЃЉдкШЫЙЄжЧФмЃЈAIЃЉЪБДњдкЛЏбЇКЭВФСЯПЦбЇжаЕФжааФзїгУЁЃDFTвђЦфНЯИпЕФдЄВтФмСІЁЂЪЪгУадЁЂЖрЙІФмадКЭМЦЫуаЇТЪЖјГЩЮЊИУСьгђЕФЙиМќЙЄОпЁЃЮФеТжиЕуЬжТлСЫЛљгкDFTЕФЛњЦїбЇЯАЃЈMachine Learning, MLЃЉФЃаЭЕФзюаТНјеЙЃЌетаЉФЃаЭдкКЯГЩЪ§ОнЩњГЩКЭФЃаЭМмЙЙЩшМЦЗНУцбЯживРРЕDFTЁЃетаЉЗЂеЙЖдЛЏбЇКЭВФСЯПЦбЇЕФвЛАуЯрЙиадНјааСЫИќЙуЗКЕФЬжТлЁЃЛљгкDFTЕФMLФЃаЭвбОЪЕЯжСЫИпаЇТЪЁЂзМШЗадЁЂПЩРЉеЙадКЭПЩзЊвЦадЃЌВЂЮЊздЖЏЪЕбщЪвжаГЩЙІЪЕбщМЦЛЎШэМўЕФГЃЙцЪЙгУЦЬЦНСЫЕРТЗЁЃ
БГОАНщЩм
ЮвУЧЩњЛюдкШЫЙЄжЧФмЪБДњЃЌAIвбОДЅМАВЂгАЯьСЫШЫРрЛюЖЏЕФМИКѕУПИіСьгђЁЃР§ШчЃЌдкздШЛгябдДІРэЁЂМЦЫуЛњЪгОѕКЭдЄВтЕШСьгђеМОнСЫжааФЮшЬЈЁЃДѓдМ20ФъЧАЃЌвЛИіУћЮЊЁАAdamЁБЕФЛљгкAIЕФЛњЦїШЫПЦбЇМвБЛв§ШыЕНКЯГЩЩњЮябЇСьгђЃЌвдажњКЭМгЫйПЦбЇЗЂЯжЁЃНќФъРДЃЌЛЏбЇКЭВФСЯЕФЛњЦїШЫКЭзджїЪЕбщШЁЕУСЫгаЯЃЭћЕФГѕВНГЩЙћЃЌР§ШчЮЊБЁФЄЗЂЯжЬсЙЉздЖЏМнЪЛЪЕбщЪвЁЃетаЉЭЛЦЦадНјеЙвбОДйЪЙШЫУЧжиаТПМТЧПЦбЇЙ§ГЬжаЁАРэНтЁБЕФКЌвхЁЃ
ЭМЮФНтЮі
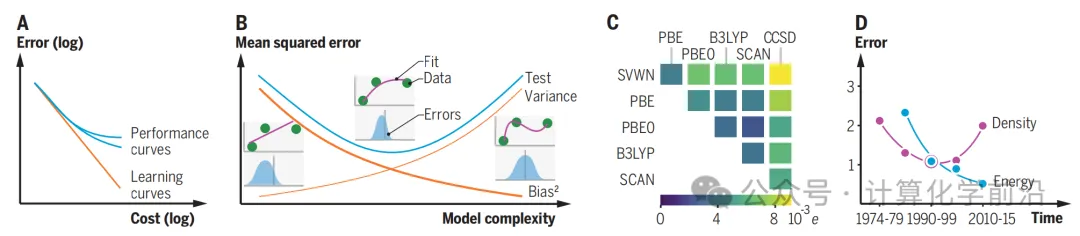
ЭМ1 еЙЪОСЫMLФЃаЭУцСйЕФЬєеНЃЌАќРЈдЄВтЮѓВюЫцзХбЕСЗМЏДѓаЁЕФдіМгЖјЫЅМѕЕФЙцТЩЃЌвдМАадФмЧњЯпВЂВЛвЛЖЈЛсЫцзХЫ№ЪЇКЏЪ§жаЮДУїШЗАќКЌЕФЯрЙижИБъЕФЬсИпЖјЬсИпЁЃЭМжаЛЙеЙЪОСЫВтЪдЮѓВюЕФзщКЯЃЌАќРЈФЃаЭЪЪгІЪ§ОнЕуЕФЦЋВюКЭФЃаЭдкЪ§ОнЕужЎМфЕФСщЛюадв§Ц№ЕФЗНВюЁЃ
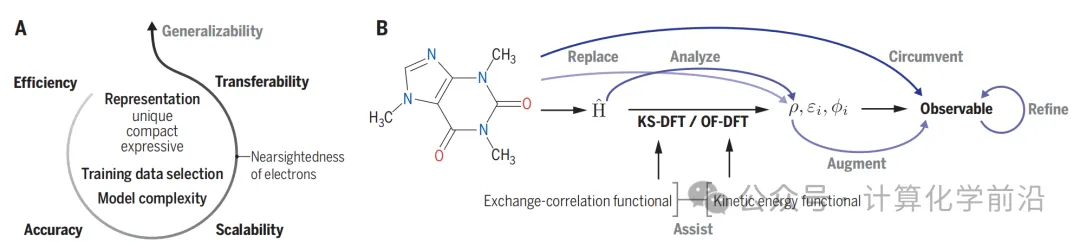
ЭМ2 ЬсЙЉСЫDFTдкMLФЃаЭжаЫљАчбнЕФЙиМќНЧЩЋЕФИХФюадИХЪіЁЃЫќИљОнаЇТЪЃЈEЃЉЁЂзМШЗадЃЈAЃЉЁЂПЩРЉеЙадЃЈSЃЉКЭПЩзЊвЦадЃЈTЃЉЫФИіЙиМќРрБ№РДЩѓВщЛЏбЇКЭВФСЯЪєадМАЙ§ГЬЕФдЄВтMLФЃаЭЁЃЭМ2ЛЙеЙЪОСЫГЌдНDFTЕФMLЗНЗЈЃЌАќРЈЭЈЙ§беЩЋжИЪОЕФЛљгкDFTЕФДЋЭГЙЄзїСїГЬКЭMLВЩгУЕФТЗЯпЁЃ
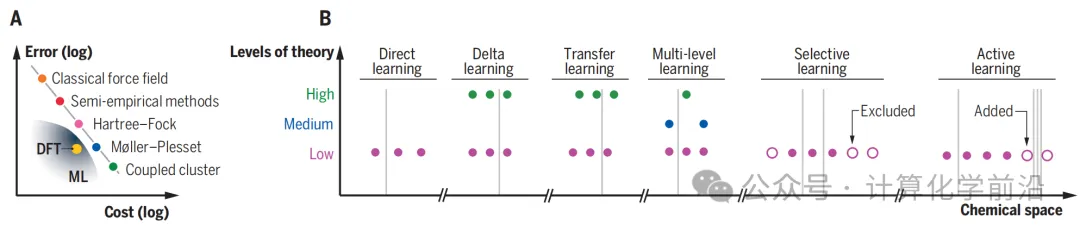
ЭМ3 УшЪіСЫдкЛЏбЇПеМфжаВЩбљЪБЕФдЄЫуИажЊМЦЫуВпТдЃЌАќРЈВЛЭЌМЖБ№ЕФРэТлЁЂбЁдёадбЇЯАЁЂЖрМЖбЇЯАЁЂжБНгбЇЯАЁЂдіСПбЇЯАКЭжїЖЏбЇЯАЕШВпТдЁЃетаЉВпТджМдкЬсИпЪ§ОнаЇТЪЃЌЭЈЙ§ВЛЭЌбеЩЋБэЪОВЛЭЌМЖБ№ЕФРэТлЪ§ОнЕуЃЌЛвЩЋЯпБэЪОаТЛЏКЯЮяЕФВщбЏЁЃ
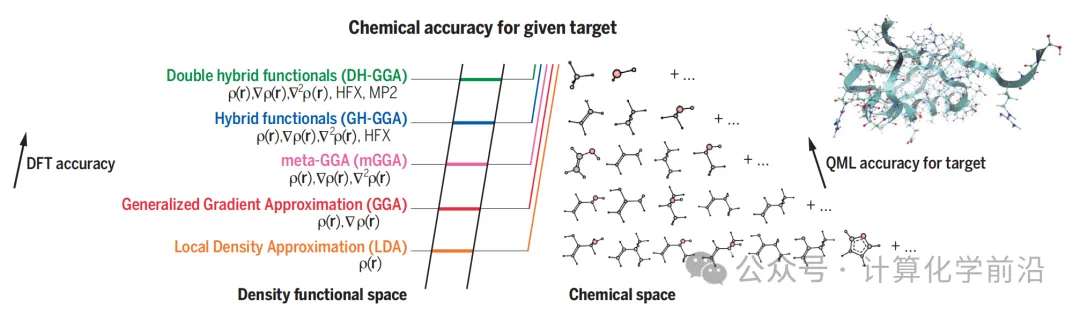
ЭМ4 еЙЪОСЫЁАбХИїВМЬнзгЁБЃЈJacobЁЏs ladderЃЉЃЌетЪЧжИЭЈГЃгУРДФЃФтЮДжЊЕФОЋШЗНЛЛЛ-ЯрЙиЪЦЕФВуДЮНсЙЙЁЃЭМжаЛЙеЙЪОСЫШчКЮЪЙгУCQMLКЭЦЌЖЮПеМфЕФВуДЮНсЙЙЃЈР§ШчamonЃЉРДдЄВтДѓаЭВщбЏФПБъЃЈШчВхЭМЫљЪОЕФаЁЕААзЗКЫиЃЉЕФЪєадЁЃ
змНсеЙЭћ
ЮФеТзмНсСЫDFTдкQMLФЃаЭЕФЗЂеЙжаЫљАчбнЕФЙЄОпадНЧЩЋЃЌетаЉФЃаЭФмЙЛЪЙгУEASTЃЈаЇТЪЁЂзМШЗадЁЂПЩРЉеЙадЁЂПЩзЊвЦадЃЉЕМКНЛЏбЇЛЏКЯЮяПеМфЁЃDFTВЛНіЪЧСПзгСІбЇНќЫЦЗНЗЈЕФЛљДЁРэТлЛљДЁЃЌЖјЧвзїЮЊГіЩЋЕФМЦЫуЪєадРДдДЃЌОпгаПЩПиЕФКЯРэЛёШЁГЩБОКЭзюЪмЛЖгЕФзМШЗадЁЃЮФеТШЯЮЊЃЌЛљгкDFTЕФQMLФЃаЭЕФПЩгУадЁЂЖрбљадКЭИпОЋЖШЃЈДяЕНЛђГЌЙ§ЪЕбщЫЎЦНЃЉЕФВФСЯКЭЗжзгЪєадЪ§ОнМЏЕФЗЂеЙКЭбЕСЗНЋЪЧЪЕЯжПЩЦеБщДІРэШЮКЮЪєадКЭЛЏбЇЕФПЩзЊвЦQMLФЃаЭЕФЛљБОвЊЧѓЁЃзюКѓЃЌЮФеТеЙЭћСЫЛљгкЮяРэЕФQMLФЃаЭгыЛњЦїШЫгВМўКЭЩшБИЕФЮоЗьМЏГЩЃЌетНЋЪЧЪЕЯжЪЕбщЪвИїжжЪЕбщШЮЮёЕФЙиМќЃЌдЄЪОзХВЛОУЕФНЋРДНЋГіЯжздЖЏМнЪЛЪЕбщЪвЁЃ