вЛЃЌМђНщ
ЗжзгФЃФтЪЧвЛжжЧПДѓЕФМЦЫуЙЄОпЃЌЫќПЩвдЭЈЙ§ФЃФтЗжзгКЭдзгЕФдЫЖЏКЭЯрЛЅзїгУРДбаОПЮяжЪЕФНсЙЙКЭааЮЊЁЃетжжЗНЗЈдкЛЏбЇЁЂЮяРэбЇКЭЩњЮябЇжаОпгаЙуЗКЕФгІгУЃЌШчбаОПЕААзжЪелЕўЁЂвЉЮяЩшМЦЁЂВФСЯПЦбЇКЭЛЏбЇЗДгІЛњжЦЕШЁЃЗжзгФЃФтЕФзМШЗаддкКмДѓГЬЖШЩЯвРРЕгкЫљЪЙгУЕФФЃаЭКЭВЮЪ§ЃЌЖјЫЎФЃаЭдкЦфжаАчбнСЫжСЙиживЊЕФНЧЩЋЁЃ
ЫЎдкздШЛНчжаЮоДІВЛдкЃЌЪЧЩњУќЛюЖЏКЭаэЖрЛЏбЇЙ§ГЬЕФживЊНщжЪЁЃгЩгкЫЎЕФЗжзгНсЙЙКЭЧтМќЭјТчЃЌЪЙЦфОпгаЖРЬиЕФЮяРэКЭЛЏбЇаджЪЃЌвђДЫдкЗжзгФЃФтжазМШЗЕиФЃФтЫЎЕФааЮЊЯдЕУгШЮЊживЊЁЃШЛЖјЃЌгЩгкЫЎЗжзгМфЕФИДдгЯрЛЅзїгУЃЌвдМАЫЎдкВЛЭЌЛЗОГжаЕФВЛЭЌБэЯжЃЌЙЙНЈвЛИіМШФмзМШЗЗДгГЫЎЕФКъЙладжЪгжФмОЋШЗУшЪіЮЂЙлааЮЊЕФЫЎФЃаЭвЛжБЪЧвЛИіОоДѓЕФЬєеНЁЃ
БОЮФжМдкЬНЬжЫЎФЃаЭЕФЦ№дДгыЗЂеЙЃЌвдМАШмМСЛЏЕФЛљБОдРэЁЃЭЈЙ§ЛиЙЫЫЎФЃаЭЕФЗЂеЙРњЪЗЃЌЮвУЧПЩвдСЫНтЦфбнБфЙ§ГЬКЭВЛЭЌФЃаЭЕФЬиЕугыгІгУЁЃДЫЭтЃЌШмМСЛЏзїЮЊвЛжжживЊЕФЛЏбЇЙ§ГЬЃЌЙуЗКгАЯьзХШмжЪдкШмМСжаЕФааЮЊЃЌРэНтЦфдРэЖдзМШЗФЃФтЩњЮяЗжзгКЭВФСЯЕФааЮЊжСЙиживЊЁЃ
ЖўЃЌЫЎФЃаЭЕФЦ№дДгыЗЂеЙ
2.1 дчЦкЫЎФЃаЭ
Lennard-JonesФЃаЭ[1]
Lennard-JonesФЃаЭЪЧзюдчЦкЕФЗжзгМфЯрЛЅзїгУФЃаЭжЎвЛЃЌзюГѕгУгкУшЪіЖшадЦјЬхдзгЕФЯрЛЅзїгУЁЃИУФЃаЭВЩгУСЫLennard-JonesЪЦКЏЪ§ЃЌЖЈвхСЫЗжзгМфЕФЮќв§КЭХХГтСІЁЃОЁЙметжжФЃаЭМђЕЅЧвМЦЫуаЇТЪИпЃЌЕЋЫќЮоЗЈзМШЗВЖзНЫЎЗжзгМфИДдгЕФЧтМќЯрЛЅзїгУЃЌвђДЫдкУшЪіЫЎЕФаджЪЗНУцДцдкЯджјОжЯоадЁЃ
Bernal-FowlerФЃаЭ[2]
BernalКЭFowlerдк1933ФъЬсГіСЫвЛжждчЦкЕФЫЎЗжзгФЃаЭЃЌГЂЪдЭЈЙ§ПМТЧЫЎЗжзгЕФЫФУцЬхНсЙЙРДНтЪЭЦфЖРЬиЕФЮяРэаджЪЁЃЫћУЧЕФФЃаЭЪзДЮв§ШыСЫЧтМќЕФИХФюЃЌЫфШЛетвЛФЃаЭдкММЪѕЩЯШдШЛгаЯоЃЌЕЋЮЊКѓајИќИДдгКЭОЋШЗЕФЫЎФЃаЭЕьЖЈСЫЛљДЁЁЃ
2.2 ОЕфЫЎФЃаЭЕФГіЯж
SPCЃЈSimple Point ChargeЃЉФЃаЭ[3]
SPCФЃаЭгЩBerendsenЕШШЫдк1981ФъЬсГіЃЌЪЧвЛжжЙуЗКЪЙгУЕФШ§ЕуФЃаЭЁЃИУФЃаЭНЋЫЎЗжзгМђЛЏЮЊШ§ИіДјЕчЕуЃЌЦфжаСНИіЕуДњБэЧтдзгЃЌвЛИіЕуДњБэбѕдзгЃЌЧвЧтбѕМќГЄКЭHOHНЧЙЬЖЈЁЃSPCФЃаЭФмЙЛдкНЯЕЭЕФМЦЫуГЩБОЯТЬсЙЉЖдвКЬЌЫЎКъЙладжЪЕФКЯРэУшЪіЃЌЕЋдквЛаЉЯИЮЂаджЪЩЯШдгаВЛзуЁЃ
TIP3PЃЈTransferable Intermolecular Potential with 3 PointsЃЉФЃаЭ[4]
TIP3PФЃаЭЪЧгЩJorgensenЕШШЫдк1983ФъПЊЗЂЕФШ§ЕуЫЎФЃаЭЁЃгыSPCФЃаЭЯрЫЦЃЌTIP3PФЃаЭЭЌбљНЋЫЎЗжзгМђЛЏЮЊШ§ИіЕуЕчКЩЃЌЕЋЦфВЮЪ§ОЙ§гХЛЏЃЌФмЙЛИќКУЕидйЯжЫЎЕФУмЖШКЭОЖЯђЗжВМКЏЪ§ЁЃTIP3PФЃаЭдкЗжзгЖЏСІбЇФЃФтжаБЛЙуЗКЪЙгУЃЌгШЦфЪЧдкЩњЮяЗжзгЕФбаОПжаЁЃ
TIP4P[5]КЭTIP5PФЃаЭ[6]
TIP4PФЃаЭгЩJorgensenЕШШЫдк1983ФъЬсГіЃЌЪЧTIP3PФЃаЭЕФИФНјАцЁЃИУФЃаЭдіМгСЫвЛИіащФтЕФMЕуЃЌвдИќКУЕиФЃФтЫЎЗжзгЕФЕчКЩЗжВМЃЌДгЖјИќзМШЗЕиУшЪіЫЎЕФОВЕчаджЪЁЃTIP5PФЃаЭНјвЛВНдіМгСЫСНИіащФтЕуЃЌРДФЃФтбѕдзгЩЯЕФЙТЖдЕчзгЁЃетСНИіФЃаЭдкФЃФтЫЎЕФНсЙЙКЭЖЏСІбЇаджЪЗНУцБэЯжИќКУЃЌЕЋМЦЫуГЩБОЯрЖдНЯИпЁЃ
2.3 ЯжДњЫЎФЃаЭЕФЗЂеЙ
POL3ЃЈPolarizable 3-site water modelЃЉ[7]
POL3ФЃаЭв§ШыСЫМЋЛЏаЇгІЃЌЪЙЕУФЃаЭФмЙЛЖЏЬЌЕїећЫЎЗжзгЕФЕчКЩЗжВМЁЃетжжМЋЛЏФЃаЭФмЙЛИќзМШЗЕиУшЪіЫЎдкВЛЭЌЛЗОГЯТЕФааЮЊЃЌЬиБ№ЪЧдкЧПЕчГЁЛђНчУцЧщПіЯТЁЃ
SPC/EЃЈExtended Simple Point ChargeЃЉФЃаЭ[8]
SPC/EФЃаЭЪЧЖдSPCФЃаЭЕФРЉеЙЃЌгЩBerendsenЕШШЫдк1987ФъЬсГіЁЃИУФЃаЭдкБЃГжМЦЫуаЇТЪЕФЭЌЪБЃЌЭЈЙ§в§ШыЖюЭтЕФФмСПаое§ЯюЃЌЬсИпСЫЖдЫЎЕФУмЖШЃЌНщЕчГЃЪ§КЭздРЉЩЂЯЕЪ§ЕФУшЪіОЋЖШЁЃ
AMOEBAЃЈAtomic Multipole Optimized Energetics for Biomolecular ApplicationsЃЉФЃаЭ[9]
AMOEBAФЃаЭгЩPonderЕШШЫПЊЗЂЃЌЪЧвЛжжЖрМЋзгФЃаЭЃЌФмЙЛзМШЗЕиУшЪіЗжзгМфИДдгЕФЕчКЩЯрЛЅзїгУЁЃИУФЃаЭдкФЃФтЩњЮяЗжзгКЭШмвКЬхЯЕжаБэЯжГіЩЋЃЌЕЋМЦЫуИДдгЖШНЯИпЁЃ
ЭЈЙ§етаЉЫЎФЃаЭЕФЗЂеЙЃЌЮвУЧПЩвдПДЕНУПИіФЃаЭЖМЪЧЖдЧАШЫЙЄзїЕФИФНјКЭгХЛЏЃЌж№ВНЬсИпСЫЖдЫЎЕФИДдгааЮЊЕФФЃФтОЋЖШЁЃОЁЙмШчДЫЃЌПЊЗЂвЛИіФмЙЛдкЫљгаЬѕМўЯТЖМОЋШЗУшЪіЫЎЕФЭЈгУФЃаЭШдШЛЪЧвЛИіОоДѓЕФЬєеНЁЃ
Ш§ЃЌЫЎФЃаЭЕФЙЙНЈЗНЗЈ
3.1 ВЮЪ§ЛЏЗНЗЈ
ЫЎФЃаЭЕФЙЙНЈЭЈГЃЩцМАВЮЪ§ЛЏЙ§ГЬЃЌЦфжаАќРЈОбщВЮЪ§ЛЏКЭАыОбщВЮЪ§ЛЏЗНЗЈЁЃ
ОбщВЮЪ§ЛЏ
ОбщВЮЪ§ЛЏЗНЗЈвРРЕгкЪЕбщЪ§ОнКЭвбжЊЕФЗжзгаджЪЁЃбаОПШЫдБЭЈЙ§ЕїећФЃаЭВЮЪ§ЃЌЪЙФЃФтНсЙћгыЪЕбщЪ§ОнЯрЮЧКЯЁЃР§ШчЃЌSPCКЭTIP3PФЃаЭЕФВЮЪ§ЪЧЭЈЙ§ФтКЯвКЬЌЫЎЕФУмЖШЁЂОВЕчаджЪКЭЯрБфааЮЊЕШЪЕбщЪ§ОнЕУЕНЕФЁЃ
АыОбщВЮЪ§ЛЏ
АыОбщВЮЪ§ЛЏЗНЗЈНсКЯСЫРэТлМЦЫуКЭЪЕбщЪ§ОнЁЃДЫЗНЗЈРћгУМђЛЏЕФСПзгЛЏбЇМЦЫуРДЛёЕУГѕЪМВЮЪ§ЃЌШЛКѓЭЈЙ§гыЪЕбщЪ§ОнЖдБШНјааЕїећЁЃTIP4PКЭTIP5PФЃаЭОЭЪЧЪЙгУетжжЗНЗЈгХЛЏЕУРДЕФЃЌвдИќКУЕиФЃФтЫЎЗжзгЕФЕчКЩЗжВМКЭЧтМќЭјТчЁЃ
3.2 ЛљгкСПзгЛЏбЇМЦЫуЕФЗНЗЈ
СПзгЛЏбЇМЦЫуЗНЗЈдкЫЎФЃаЭЕФЙЙНЈжавВАчбнСЫживЊНЧЩЋЃЌЬиБ№ЪЧдкМЋЛЏФЃаЭКЭЖрМЋзгФЃаЭЕФПЊЗЂжаЁЃ
ДгЭЗМЦЫу
ДгЭЗМЦЫуЃЈAb initioМЦЫуЃЉЪЧЛљгкСПзгСІбЇдРэЕФЕквЛаддРэМЦЫуЗНЗЈЁЃЭЈЙ§ДгЭЗМЦЫуЃЌПЩвдЛёЕУЫЎЗжзгЕФЕчзгЗжВМКЭЪЦФмУцЁЃPOL3КЭAMOEBAФЃаЭРћгУДгЭЗМЦЫуЕУЕНЕФЗжзгМфЯрЛЅзїгУаХЯЂРДЙЙНЈФЃаЭВЮЪ§ЁЃ
ЗжзгСІГЁЕФЙЙНЈ
ЗжзгСІГЁЕФЙЙНЈЩцМАЖЈвхЗжзгМфЯрЛЅзїгУЕФЪЦКЏЪ§ЃЌАќРЈЕчКЩЗжВМЁЂМќГЄЁЂМќНЧКЭХЄзЊНЧЕШЁЃСПзгЛЏбЇМЦЫуЬсЙЉСЫетаЉВЮЪ§ЕФГѕЪМЙРМЦжЕЃЌЖјКѓајЕФВЮЪ§гХЛЏЙ§ГЬЪЙФЃаЭФмЙЛИќзМШЗЕиЗДгГЪЕбщЙлВтЕНЕФЮяРэаджЪЁЃ
3.3 ЪЕбщЪ§ОнЕФаЃзМ
ЪЕбщЪ§ОндкЫЎФЃаЭЕФЙЙНЈКЭбщжЄЙ§ГЬжажСЙиживЊЁЃ
ЪЙгУЪЕбщЪ§ОнНјааВЮЪ§гХЛЏ
ЫЎФЃаЭЕФВЮЪ§ЭЈГЃашвЊЭЈЙ§гыЪЕбщЪ§ОнЕФЖдБШНјаааЃзМЁЃР§ШчЃЌЫЎЕФНщЕчГЃЪ§ЁЂздРЉЩЂЯЕЪ§КЭШШШнЕШКъЙладжЪЪЧГЃгУЕФаЃзМБъзМЁЃSPC/EФЃаЭОЭЪЧЭЈЙ§в§ШыФмСПаое§ЯюЃЌЪЙФЃаЭИќКУЕидйЯжЫЎЕФНщЕчГЃЪ§ЁЃ
ФЃФтгыЪЕбщНсЙћЕФЖдБШ
ЙЙНЈКУЕФЫЎФЃаЭашвЊЭЈЙ§гыЪЕбщНсЙћЕФЖдБШРДбщжЄЦфзМШЗадЁЃФЃФтНсЙћгІФмЙЛжиЯжЪЕбщжаЙлВьЕНЕФЫЎЕФЮяРэКЭЛЏбЇаджЪЃЌШчвКЬЌЫЎЕФНсЙЙЁЂЖЏСІбЇаджЪКЭЯрБфааЮЊЁЃетжжЖдБШВЛНібщжЄСЫФЃаЭЕФзМШЗадЃЌЛЙФмжИЕМНјвЛВНЕФФЃаЭИФНјЁЃ
ЭЈЙ§ЩЯЪіЗНЗЈЃЌбаОПШЫдБФмЙЛЙЙНЈГідНРДдНОЋШЗЕФЫЎФЃаЭЃЌвдТњзуВЛЭЌПЦбЇбаОПЕФашЧѓЁЃ
ЫФЃЌШмМСЛЏ
4.1 ШмМСЛЏЕФЖЈвх
дкЯъЯИЬНЬжСЫЫЎФЃаЭЕФЦ№дДгыЙЙНЈЗНЗЈжЎКѓЃЌЮвУЧНгЯТРДНЋжиЕуНщЩмШчКЮдкЗжзгФЃФтжагІгУетаЉЫЎФЃаЭРДНјааШмМСЛЏЙ§ГЬЁЃШмМСЛЏЙ§ГЬВЛНіЪЧЗжзгФЃФтжаВЛПЩЛђШБЕФвЛВПЗжЃЌвВЪЧРэНтЗжзгааЮЊКЭЯрЛЅзїгУЕФЙиМќЁЃЭЈЙ§ШмМСЛЏЃЌбаОПШЫдБФмЙЛдкФЃФтжаИќецЪЕЕидйЯжЪЕМЪЛЗОГжаЕФЗжзгааЮЊЃЌДгЖјЬсИпФЃФтНсЙћЕФзМШЗадКЭПЩППадЁЃ
ШмМСЛЏЃЈSolvationЃЉЪЧжИШмжЪЗжзгдкШмМСжаБЛШмМСЗжзгАќЮЇКЭЮШЖЈЕФЙ§ГЬЁЃетИіЙ§ГЬдкЛЏбЇЁЂЮяРэбЇКЭЩњЮябЇжаОпгаживЊвтвхЃЌвђЮЊаэЖрЛЏбЇЗДгІЁЂЩњЮяЙ§ГЬКЭВФСЯааЮЊЖМЗЂЩњдкШмвКжаЁЃЬиБ№ЪЧдкЗжзгФЃФтжаЃЌЭЈЙ§ЖдЬхЯЕНјааШмМСЛЏЃЌбаОПШЫдБПЩвдИќзМШЗЕиФЃФтЩњЮяЗжзгЁЂвЉЮяЗжзгвдМАЦфЫћЛЏбЇЯЕЭГдкЪЕМЪЛЗОГжаЕФааЮЊЁЃ
4.2 ШмМСЛЏЕФживЊад
ШмМСЛЏЙ§ГЬЩцМАШмМСЗжзгЮЇШЦШмжЪЗжзгаЮГЩШмМСПЧВуЕФЙ§ГЬЁЃетИіПЧВуЭЈЙ§ИїжжЗжзгМфЯрЛЅзїгУЃЈШчЧтМќЁЂЗЖЕТЛЊСІЁЂЕчКЩЯрЛЅзїгУЕШЃЉЮШЖЈШмжЪЗжзгЁЃР§ШчЃЌдкЫЎзїЮЊШмМСЕФЧщПіЯТЃЌЫЎЗжзгЭЈЙ§ЧтМќКЭХММЋЯрЛЅзїгУЃЌгыШмжЪЗжзгаЮГЩЮШЖЈЕФНсЙЙЁЃетжжШмМСЛЏПЧВуВЛНігАЯьШмжЪЗжзгЕФЮШЖЈадЃЌЛЙгАЯьЦфЖЏСІбЇаджЪКЭЗДгІЛњжЦЁЃвђДЫШмМСЛЏЙ§ГЬЖдШмжЪЗжзгЕФааЮЊгазХЙуЗКЕФгАЯьЃЌдквдЯТМИИіЗНУцгШЮЊживЊЃК
ЮШЖЈадгыШмНтадЃКШмМСЛЏЙ§ГЬЯджјгАЯьШмжЪЗжзгЕФЮШЖЈадКЭШмНтадЁЃЭЈЙ§ШмМСЛЏЃЌШмжЪЗжзгФмЙЛдкШмМСжаБЃГжЮШЖЈЃЌЗРжЙОлМЏЛђГСЕэЁЃР§ШчЃЌЕААзжЪдкЫЎжаЕФШмНтКЭЮШЖЈадЪЧгЩЦфгыЫЎЗжзгЕФШмМСЛЏЯрЛЅзїгУОіЖЈЕФЁЃ
ЗДгІЖЏСІбЇЃКШмМСЛЏЛЙгАЯьЛЏбЇЗДгІЕФЖЏСІбЇаджЪЁЃШмМСЗжзгЭЈЙ§ЮШЖЈЗДгІжаМфЬхКЭЙ§ЖЩЬЌЃЌНЕЕЭЗДгІЛюЛЏФмЃЌДгЖјМгЫйЗДгІЫйТЪЁЃР§ШчЃЌУИДйЗДгІжаЃЌЫЎЗжзгВЛНізїЮЊШмМСЃЌЛЙВЮгыСЫУИгыЕзЮяЕФЯрЛЅзїгУКЭЗДгІЙ§ГЬЁЃ
ЩњЮяЗжзгЙІФмЃКЩњЮяДѓЗжзгЃЈШчЕААзжЪЁЂКЫЫсЕШЃЉдкЩњРэЬѕМўЯТЕФНсЙЙКЭЙІФмвРРЕгкШмМСЛЏЛЗОГЁЃЫЎЗжзгЭЈЙ§гыЩњЮяДѓЗжзгЕФЧтМќКЭЪшЫЎЯрЛЅзїгУЃЌгАЯьЦфелЕўЁЂЙЙЯѓБфЛЏКЭЙІФмБэЯжЁЃР§ШчЃЌЕААзжЪЕФЛюадЮЛЕуГЃГЃАќКЌвЛВуЬиЖЈХХСаЕФЫЎЗжзгЃЌетаЉЫЎЗжзгдкУИДйЗДгІжаЦ№зХЙиМќзїгУЁЃ
ВФСЯПЦбЇЃКдкВФСЯПЦбЇжаЃЌШмМСЛЏЙ§ГЬгАЯьВФСЯЕФКЯГЩЁЂМгЙЄКЭадФмЁЃР§ШчЃЌОлКЯЮяШмвКЕФеГЖШКЭСїБфаджЪвРРЕгкОлКЯЮяСДгыШмМСЗжзгЕФЯрЛЅзїгУЁЃЭЈЙ§РэНтКЭПижЦШмМСЛЏЙ§ГЬЃЌПЩвдгХЛЏВФСЯЕФадФмКЭгІгУЁЃ
вЉЮяЩшМЦгыПЊЗЂЃКдквЉЮяЩшМЦжаЃЌШмМСЛЏЖдвЉЮяЗжзгЕФЮќЪеЁЂЗжВМЁЂДњаЛКЭХХаЙЃЈADMEЃЉгаживЊгАЯьЁЃвЉЮяЗжзггыЫЎЗжзгЕФЯрЛЅзїгУгАЯьЦфШмНтЖШКЭЩњЮяРћгУЖШЁЃЭЈЙ§ЗжзгФЃФтбаОПШмМСЛЏЙ§ГЬЃЌПЩвдгХЛЏвЉЮяЗжзгЕФНсЙЙЃЌЬсИпЦфвЉаЇКЭАВШЋадЁЃ
злЩЯЫљЪіЃЌШмМСЛЏЙ§ГЬдкЖржжПЦбЇСьгђжаОпгаживЊЕФгІгУМлжЕЁЃЭЈЙ§ЗжзгФЃФтжаЕФШмМСЛЏВйзїЃЌбаОПШЫдБПЩвдИќКУЕиРэНтКЭдЄВтШмжЪЗжзгдкШмвКжаЕФааЮЊЃЌЮЊПЦбЇбаОПКЭгІгУЬсЙЉЧПгаСІЕФжЇГжЁЃ
4.3 ШмМСЛЏЕФГЃМћЗНЗЈ
дкЗжзгФЃФтжаЃЌгаМИжжГЃМћЕФЗНЗЈгУгкЖдЬхЯЕНјааШмМСЛЏЃК
КазгЗНЗЈЃЈBox MethodЃЉ
КазгЗНЗЈЪЧзюГЃгУЕФШмМСЛЏЗНЗЈжЎвЛЁЃдкетжжЗНЗЈжаЃЌЪзЯШЖЈвхвЛИіШ§ЮЌФЃФтКаЃЌШЛКѓНЋШмжЪЗжзгЗХжУдкКазгЕФжабыЃЌНгзХЭЈЙ§ЬюГфШмМСЗжзгРДЭъГЩШмМСЛЏЁЃ
ВНжшЃК
ЖЈвхФЃФтКаЕФГпДчЃКШЗЖЈФЃФтКаЕФДѓаЁЭЈГЃашвЊШЗБЃШмжЪЗжзггыБпНчжЎМфгазуЙЛЕФОрРыЃЌвдБмУтБпНчаЇгІЕФгАЯьЁЃвЛАуЧщПіЯТЃЌжСЩйашвЊдкШмжЪЗжзгжмЮЇБЃСє1.0ФЩУзЕФПеМфЁЃ
ЗХжУШмжЪЗжзгдкКазгжааФЃКНЋШмжЪЗжзгЃЈШчЕААзжЪЛђаЁЗжзгЃЉЗХжУдкФЃФтКаЕФжааФЃЌвдШЗБЃЦфБЛШмМСЗжзгОљдШАќЮЇЁЃ
ЬэМгШмМСЗжзгЃКЭЈЙ§ЬюГфШмМСЗжзгЃЈШчЫЎЗжзгЃЉРДЬюТњећИіФЃФтКаЁЃЭЈГЃЪЙгУдЄЯШЦНКтЕФШмМСФЃаЭЃЈШчSPCЛђTIP3PЫЎФЃаЭЃЉРДНјааЬюГфЁЃ
гХШБЕуЃК
гХЕуЃКМђЕЅжБНгЃЌвзгкЪЕЯжЃЌЪЪгУгкДѓЖрЪ§ЗжзгФЃФтЯЕЭГЁЃ
ШБЕуЃКПЩФмЛсГіЯжШмМСЗжзгжиЕўЛђВЛОљдШЗжВМЕФЮЪЬтЃЌашвЊКѓајДІРэЃЌШчФмСПзюаЁЛЏКЭЗжзгЖЏСІбЇЦНКтЛЏЁЃ
ЫцЛњЗХжУЗЈЃЈRandom Placement MethodЃЉ
ЫцЛњЗХжУЗЈЪЧНЋШмМСЗжзгЫцЛњЕиЗХжУдкФЃФтКажаЁЃетжжЗНЗЈПЩвдБмУтФГаЉЙцдђЗХжУЗНЗЈПЩФмв§ШыЕФЦЋВюЁЃ
ВНжшЃК
ЖЈвхФЃФтКаЕФГпДчЃКШЗЖЈФЃФтКаЕФДѓаЁЃЌвдШЗБЃШмжЪЗжзггыБпНчжЎМфгазуЙЛЕФОрРыЁЃ
ЫцЛњЗХжУШмжЪЗжзгЃКНЋШмжЪЗжзгЫцЛњЗХжУдкФЃФтКажаЃЌШЗБЃЦфжааФЮЛжУКЯРэЁЃ
ЫцЛњЗХжУШмМСЗжзгЃКЫцЛњЕиНЋШмМСЗжзгЗХжУдкФЃФтКажаЕФПеЯЖжаЃЌБмУтгыШмжЪЗжзгЗЂЩњжиЕўЁЃ
гХШБЕуЃК
гХЕуЃКБмУтСЫЙцдђЗХжУПЩФмДјРДЕФЯЕЭГЮѓВюЃЌдіМгСЫШмМСЗжзгЕФЖрбљадКЭОљдШадЁЃ
ШБЕуЃКМЦЫуИДдгЖШНЯИпЃЌПЩФмашвЊИќЖрЕФМЦЫузЪдДКЭЪБМфРДДІРэШмМСЗжзгЕФЫцЛњЗжВМЁЃ
дЄЖЈвхЭјИёЗЈЃЈPredefined Grid MethodЃЉ
дЄЖЈвхЭјИёЗЈЪЧНЋФЃФтКаЛЎЗжЮЊЙцдђЕФЭјИёЃЌШЛКѓНЋШмжЪКЭШмМСЗжзгАДееЭјИёЕуНјааЗХжУЁЃ
ВНжшЃК
ЖЈвхФЃФтКаЕФГпДчЃКШЗЖЈФЃФтКаЕФДѓаЁЃЌВЂНЋЦфЛЎЗжЮЊЙцдђЕФЭјИёЁЃ
ЛЎЗжЭјИёЃКНЋФЃФтКаЛЎЗжЮЊвЛЯЕСааЁЕФЭјИёЕуЃЌУПИіЭјИёЕуДњБэвЛИіЧБдкЕФШмМСЗжзгЗХжУЮЛжУЁЃ
АДееЭјИёЕуЗХжУШмжЪКЭШмМСЗжзгЃКНЋШмжЪЗжзгЗХжУдкЭјИёЕФжааФЮЛжУЃЌШЛКѓНЋШмМСЗжзгАДееЪЃгрЕФЭјИёЕуНјааЗХжУЁЃ
гХШБЕуЃК
гХЕуЃКОљдШЗжВМШмМСЗжзгЃЌМѕЩйжиЕўПЩФмадЃЌгажњгкПьЫйЩњГЩГѕЪМХфжУЁЃ
ШБЕуЃКашвЊзаЯИбЁдёЭјИёГпДчЃЌвдБмУтЮѓВюЃЌЧвдкИпУмЖШЧщПіЯТПЩФмВЛЙЛСщЛюЁЃ
4.4 groamcsШмМСЛЏЗНЗЈМАдРэ
етРявдКазгЗНЗЈЮЊР§згЃЌМђЪівЛЯТgromacsГЃМћЕФШмМСЛЏЬхЯЕЕФЙЙНЈ
ЪОР§1ЃКЙЙНЈДПЫЎЃЈШмМСЃЉЛЏЬхЯЕЕФЗНЗЈ
ВНжшЃК
gmx solvate -cs spc216 -o water_box.gro -box 3 3 3
ЦСФЛДђгЁаХЯЂШчЯТЃК
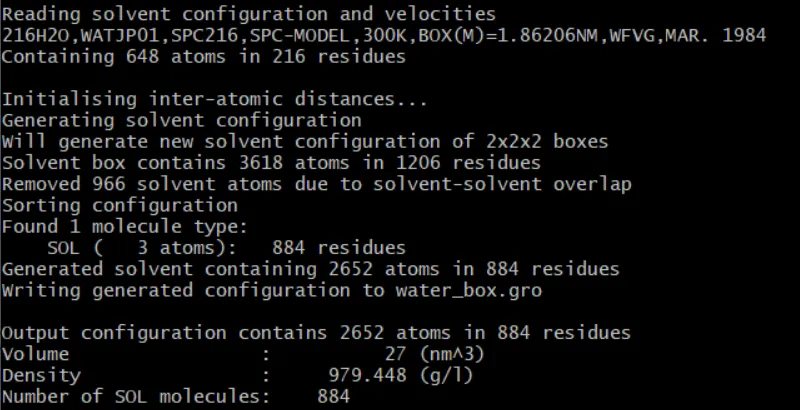
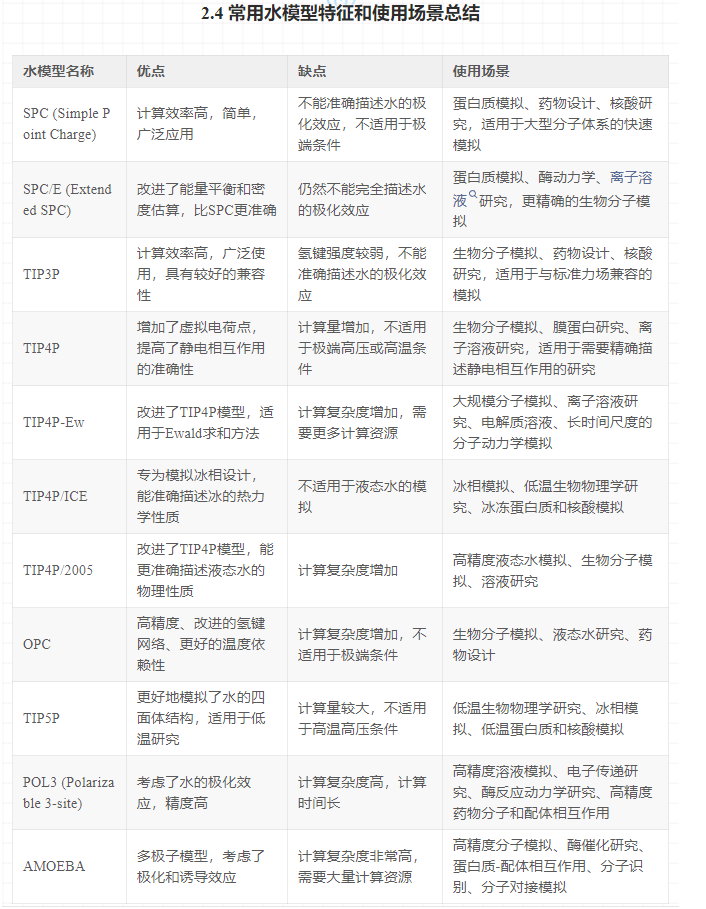
image-20240729161835494
Reading solvent configuration and velocities //ГЬађе§дкЖСШЁШмМСЕФХфжУКЭЫйЖШаХЯЂЁЃ
216H2O,WATJP01,SPC216,SPC-MODEL,300K,BOXЃЈMЃЉ=1.86206NM,WFVG,MAR. 1984ЃК//етааЯдЪОСЫдЄЖЈвхЫЎКазгЕФЯъЯИаХЯЂЁЃЫќБэУїЪЙгУЕФЪЧ216ИіЫЎЗжзгЕФSPC216ФЃаЭЃЌетЪЧЫЎЗжзгЕФвЛИіГЃМћФЃаЭЃЌЪЪгУгкдк300KЃЈПЊЖћЮФЮТЖШЃЉЯТЪЙгУЁЃBOXЃЈMЃЉБэЪОдЪМЫЎКазгЕФБпГЄЮЊ1.86206ФЩУзЁЃ
Containing 648 atoms in 216 residues //дЪМЫЎКазгАќКЌ648ИідзгЃЌЗжВМдк216ИіВаЛљжаЁЃ
Initialising inter-atomic distances... //ГЬађе§дкГѕЪМЛЏдзгМфЕФОрРыЁЃ
Generating solvent configuration //ГЬађе§дкЩњГЩШмМСЕФХфжУЁЃ
Will generate new solvent configuration of 2x2x2 boxes //ГЬађМЦЛЎЩњГЩвЛИіаТЕФШмМСХфжУЃЌЦфКазгГпДчЪЧдЪМГпДчЕФ2БЖЁЃЃЈ3nm/1.86206nmЯђЩЯШЁећЃЌXYZШ§ИіЗНЯђЃЌИїЬюГф2Иіsp216ЕФдЄЖЈгкЫЎКазгЃЉ
Solvent box contains 3618 atoms in 1206 residues //аТЕФШмМСКазгАќКЌ3618ИідзгЃЌЗжВМдк1206ИіВаЛљжаЁЃ
Removed 966 solvent atoms due to solvent-solvent overlap //гЩгкШмМСЗжзгжЎМфЕФжиЕўЃЌГЬађвЦГ§СЫ966ИіШмМСдзгЁЃ
Sorting configuration //ГЬађе§дкЖдХфжУНјааХХађЁЃ
Found 1 molecule typeЃКSOLЃЈ3 atomsЃЉЃК884 residues //ГЬађЗЂЯжСЫвЛжжЗжзгРраЭЃЌМДSOLЃЈ3ИідзгЃЉЃЌЙВга884ИіВаЛљЁЃ
Generated solvent containing 2652 atoms in 884 residues //ЩњГЩЕФШмМСАќКЌ2652ИідзгЃЌЗжВМдк884ИіВаЛљжаЁЃ
Writing generated configuration to water_box.gro //ГЬађе§дкНЋЩњГЩЕФХфжУаДШыЕНwater_box.groЮФМўжаЁЃ
Output configuration contains 2652 atoms in 884 residues //ЪфГіЕФХфжУАќКЌ2652ИідзгЃЌЗжВМдк884ИіВаЛљжаЁЃ
Volume ЃК27ЃЈnm^3ЃЉ //ЪфГіХфжУЕФЬхЛ§ЮЊ27СЂЗНФЩУзЁЃ
Density ЃК979.448ЃЈg/lЃЉ //ЪфГіХфжУЕФУмЖШЮЊ979.448ПЫ/Щ§ЁЃ
Number of SOL moleculesЃК884 //ЪфГіХфжУжаSOLЗжзгЕФЪ§СПЮЊ884ЁЃ
ШмМСЛЏЕФЙ§ГЬЃКШчЯТЭМЫљЪОЃК
❝
GROMACS ЬсЙЉМИжждЄЖЈвхЕФЫЎЗжзгХфжУЮФМўЃЌгУгкПьЫйЩшжУШмвКЛЗОГЁЃзюГЃгУЕФдЄЖЈвхЫЎКазгАќРЈЃК
spc216.groЃКетЪЧвЛИіаЁаЭЕФЫЎЗжзгКазгЃЌЭЈГЃгУгкSPCЃЈSimple Point ChargeЃЉЫЎФЃаЭЁЃетжжКазгАќКЌ216ИіЫЎЗжзгЃЌХХСаГЩвЛИіСЂЗНЬхНсЙЙЁЃ
tip4p.groЃКгУгкTIP4PЫЎФЃаЭЕФХфжУЮФМўЁЃАќКЌ216ИіЫЎЗжзгЃЌХХСаГЩвЛИіСЂЗНЬхНсЙЙЁЃ
tip5p.groЃКгУгкTIP4PЫЎФЃаЭЕФХфжУЮФМў.АќКЌ512ИіЫЎЗжзгЃЌХХСаГЩвЛИіСЂЗНЬхНсЙЙЁЃ
етаЉдЄЖЈвхКазгЭЈГЃЪЧСЂЗНЬхЛђНќЫЦСЂЗНЬхЕФаЮзДЃЌвђЮЊСЂЗНЬхКазгзюШнвзНјааМИКЮИДжЦКЭДІРэЁЃ
ЪОР§2ЃКЙЙНЈШмжЪЕФЃЈЗжзгЃЉШмМСЛЏЬхЯЕЕФЗНЗЈ
ВНжшШчЯТЃК
ЃЈ1ЃЉ.зМБИЕААзжЪЗжзгЃЌетРяЪЧЪЙгУЕФ1ycr.pdbЃЈPDBID:1YCRЃЉЕФЕААзЗжзг
ЃЈ2ЃЉЖЈвхФЃФтКаЃК
ЪЙгУeditconfУќСюЖЈвхФЃФтКаЕФГпДчКЭаЮзДЁЃ
gmx editconf -f 1ycr.pdb -o protein_box.gro -d 1.0 -bt cubic
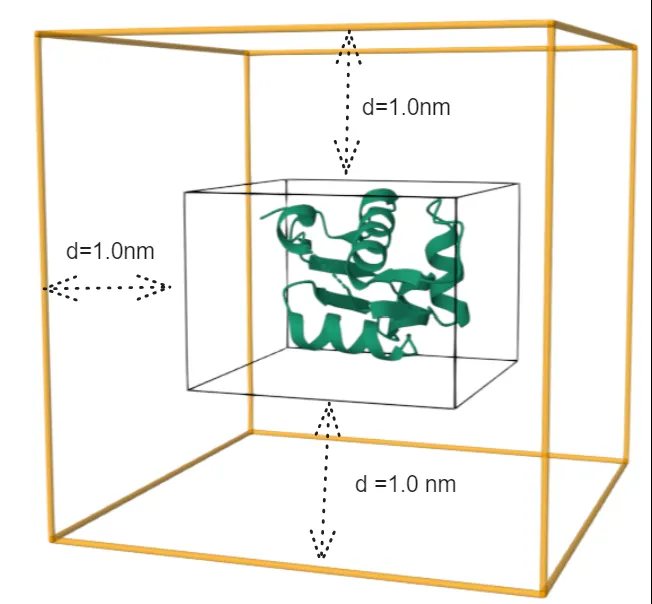
image-20240730143743113
❝
ЩЯУцЕФУќСюЪЧНЋЕААзжЪЗжзгЗХжУдкКазгЕФжаМфЃЌКазгЕФРраЭБЛЖЈвхЮЊвЛИіСЂЗНЬхЃЈ-bt cubicЃЉ,ВЂБЃГжЦфОрРыКазгБпдЕ1.0nmЃЈ-d 1.0ЃЉЁЃЕНКазгБпдЕЕФОрРыЃЈ-dЃЉ ЪЧвЛИіживЊЕФВЮЪ§ЃЌвђЮЊдкЗжзгЖЏСІбЇФЃФтЪБЃЌЮвУЧгЩгкЪЙгУСЫжмЦкадБпНчЬѕМўЃЌвђДЫашвЊТњзузюаЁЭМЯёдМЖЈЃЌМДШмжЪгРдЖВЛгІИУ"ПДЕН"ЦфжмЦкадЭМЯёЁЃетРяЮвУЧЩшжУШмжЪЃЈЕААзжЪЗжзгЃЉКЭКазгжЎМфЬэМгжСЩй 1.0 nm ЕФОрРы ЃЌетвтЮЖзХШмжЪЗжзгЕФШЮКЮСНИіжмЦкадЭМЯёжЎМфжСЩйга 2.0 nmЃЈЧызЂвтЃЌЯЕЭГжаЕФЗжзгЃЌР§ШчЕААзжЪЃЌПЩФмЛсОРњИїжжЙЙЯѓзЊБфГіЯжПчдНжмЦкадБпНчЕФЧщПіЃЉЁЃЖдгкФЃФтжаГЃгУЕФШЮКЮНижЙЗНАИЃЈЗЖЕТЛЊЃК0.8~1.2nm; ОВЕчЃК1.0~1.6nmЃЉЃЌетИіОрРыгІИУзуЙЛСЫЃЌЕЋШчЙћФуЪЙгУЕФЪЧздЖЈвхСІГЁЃЌдђПЩФмашвЊЪЙгУСІГЁТлЮФНјаабщжЄЁЃ
ЃЈ3ЃЉЬэМгШмМСЗжзгЃК
ЪЙгУsolvateУќСюНЋЫЎЗжзгЬэМгЕНФЃФтКажаЁЃ
gmx solvate -cs spc216 -cp protein_box.gro -o protein_solvate.gro
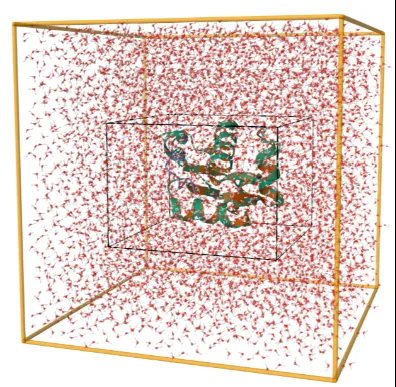
image-20240730153616818
ШмМСЛЏЕФЙ§ГЬЃКШчЯТЭМЫљЪОЃК
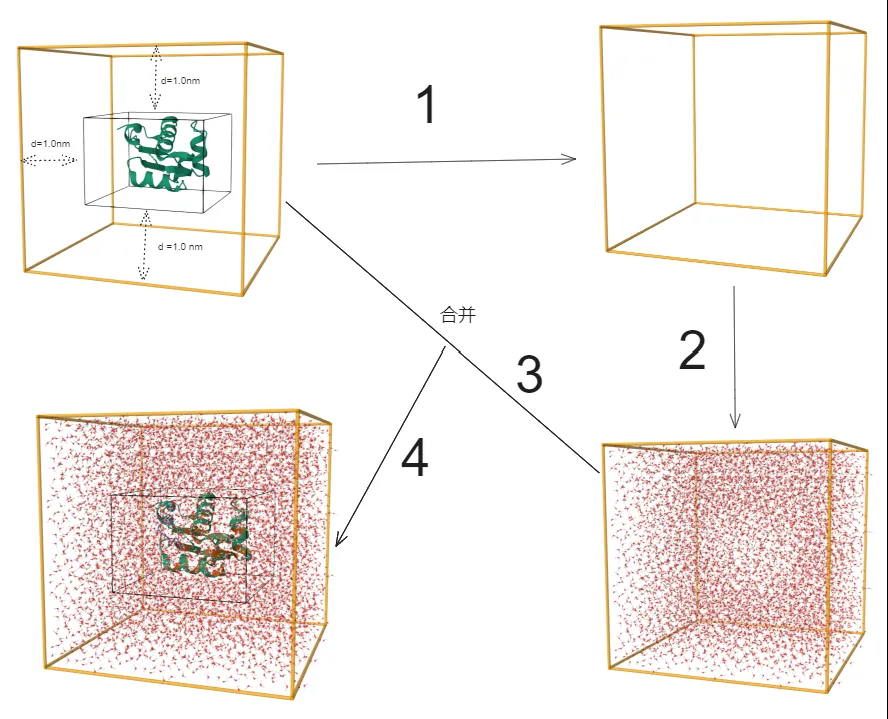
image-20240802153054166
gromacsЙЙНЈШмМСЛЏЬхЯЕЕФдРэ
ДгЩЯЪіЕФСНИіЪОР§КЭШмМСЛЏЕФЙ§ГЬжаЦСФЛДђгЁЕФаХЯЂЃЌЦфЪЕОЭПЩвджЊЕРДѓИХжЊЕРgromacsЕФШмМСЛЏЕФЛљБОдРэВНжшЕФЃЌИќМгЯъЯИЕФВНжшПЩВЮМћЦфЙигкsolvateУќСюЪЕЯжЕФдДТыЃКhttps://github.com/gromacs/gromacs/blob/2447fd95b93d00fd8472a52a6fada09eece2a42c/src/gromacs/gmxpreprocess/solvate.cpp#L861ЁЃетРяМђЕЅИХЪіЕФЯТgmx solvateКЫаФЙІФмТпМЕФЪЕЯжЃЌШчЯТЃК
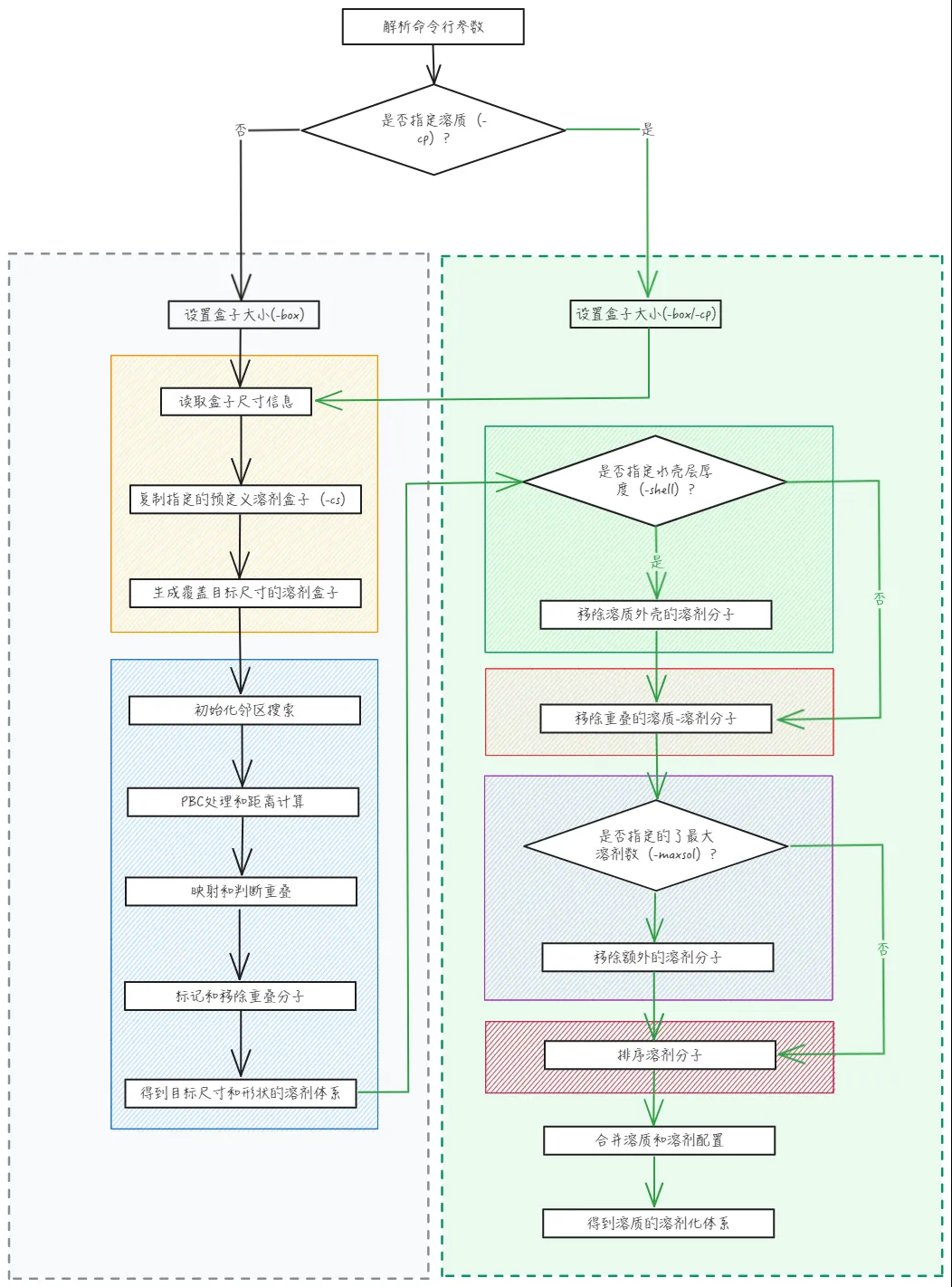
New BoardЃЈ1ЃЉ
gmx solvate зїЮЊ GROMACS ШэМўжаЕФвЛИіЙІФмЧПДѓЕФФЃПщЃЌЫќжївЊгУгкСНИіЙиМќШЮЮёЃКЩњГЩШмМСКазгКЭНјааШмжЪЕФШмМСЛЏДІРэЁЃ
ЪзЯШЃЌЫќФмЙЛИљОнгУЛЇжИЖЈЕФВЮЪ§ЃЌШчШмМСЮФМўЃЈ-csЃЉКЭКазгГпДчЃЈ-boxЃЉЃЌДДНЈвЛИіГфТњШмМСЗжзгЕФКазгЁЃетИіЙІФмЬиБ№ЪЪгУгкашвЊдЄЩшЬиЖЈГпДчШмМСЛЗОГЕФЧщПіЁЃЃЈЩЯЭМзѓВрЕФЛвЩЋБГОАЧјгђЃЉ
ЦфДЮЃЌИУФЃПщФмЙЛНЋвЛИіШмжЪЗжзгЃЌБШШчЕААзжЪЃЌШмНтдкШмМСжаЃЌаЮГЩШмМСЛЏЯЕЭГЁЃгУЛЇашвЊжИЖЈШмжЪЮФМўЃЈ-cpЃЉКЭШмМСЮФМўЃЈ-csЃЉЃЌГЬађЛсЪЙгУШмжЪзјБъЮФМўжадЄЖЈвхШмМСКазгЃЌГ§ЗЧСэЭтжИЖЈСЫКазгГпДчЃЈ-boxЃЉЁЃЃЈЩЯЭМзѓВрЛвЩЋБГОА+гвВрЧГТЬЩЋБГОАЧјгђЃЉ
дкШмМСЛЏЙ§ГЬжаЃЌgmx solvate ЛсжЧФмЕиШЅГ§гыШмжЪдзгжиЕўЕФШмМСЗжзгЃЌШЗБЃЩњГЩЕФЯЕЭГМШЗћКЯЮяРэЪЕМЪЃЌгжЪЪКЯКѓајЕФЗжзгЖЏСІбЇФЃФтЁЃ
ДЫЭтЃЌЫќЛЙЬсЙЉСЫЖдШмМСЗжзгНјааХХађЕФЙІФмЃЌвдгХЛЏФЃФтЕФаЇТЪКЭзМШЗадЁЃећИіФЃПщЕФЩшМЦПМТЧСЫгУЛЇВйзїЕФМђБуадКЭФЃФтНсЙћЕФПЩППадЃЌЪЧ GROMACS дкЗжзгФЃФтСьгђжаВЛПЩЛђШБЕФЙЄОпжЎвЛЁЃ
ЯТУцНЋМђЪіЩЯЪіСїГЬЭМжаЕФЙиМќжДааСНДѓРраЭШмМСЛЏШЮЮёЕФКЫаФЙІФмКЏЪ§ЃК
1.ЭМЦЌЃКreplicateSolventBoxЃЈЃЉЃЌИУКЏЪ§гУгкИДжЦШмМСКазгЁЃЫќЛсИљОнФПБъКЭКазгЕФДѓаЁЃЈ-boxЃЉРДМЦЫудЄЖЈвхШмМСКазгЃЈ-cs spc216ЃЉдкФПБъКазгИїЗНЯђЩЯЕФИДжЦвђзгЃЌШЛКѓдйНЋдЄЖЈШмМСКазгИДжЦЖрДЮвдЬюГфФПБъКазгЁЃетИіЙ§ГЬжаЃЌПЩФмЛсВњЩњГЌГіФПБъКазгЕФЗжзгЃЌвђЮЊФПБъКазгЕФГпДчПЩФмЮоЗЈЭъШЋБЛећЪ§БЖЕФКазгИВИЧЁЃБШШчЪОР§1жаЧщПі
2.ЭМЦЌЃКremoveSolventBoxOverlapЃЈЃЉ,ИУКЏЪ§жївЊгУгквЦГ§гЩгкдЄЖЈвхЕФШмМСКазгКЭжмЦкадБпНчЬѕМўЃЈPBCЃЉЕМжТЕФжиЕўШмМСЗжзгЁЃМђЕЅРДЫЕЦфжївЊИКд№ЕФШЮЮёЪЧ"МєВУ"ЃЌНЋreplicateSolventBoxЃЈЃЉВњЩњЕФШмМСЛЏКазг"МєВУ"ЮЊФПБъГпДчКЭаЮзДЕФШмМСЛЏЬхЯЕЁЃ
❝
ЮЊЪВУДашвЊ"МєВУ"ЃП
1.ФПБъКазгЕФГпДчПЩФмЮоЗЈЭъШЋБЛећЪ§БЖЕФдЄЖЈвхШмМСКазгИВИЧЁЃ
2.дЄЖЈвхКазгЪЧСЂЗНЬхЃЌЖјФПБъКазггаПЩФмЪЧСЂЗНЬхЃЌШ§аБОЇЯЕЃЌНиНЧАЫУцЬхЃЌСтаЮЪЎЖўУцЬхЕШЁЃ
ЮЊЪВУДВЛжБНгвЦçÍɧФПБъГпДчБпНчЕФЫЎЗжзгЃП
1.дЄЖЈвхЕФЫЎКазгФЃаЭдкЙЙНЈЪБвбОПМТЧСЫжмЦкадБпНчЬѕМўЃЌвђДЫЫќУЧБЛИДжЦЪБЖбЕўГЩИќДѓЕФШмМСЛЏКазгЪБШЗБЃдкБпНчДІЫЎЗжзгЕФЗжВМБЃГжОљдШЃЌВЂЧвВЛЛсВњЩњЖюЭтЕФУмЖШБфЛЏЃЌШчЙћДЫЪБжБНгШЅЕєБпНчЭтЕФЫЎЗжзгЃЈШчЪОР§1жаЃЌРЖЩЋПђЭтЕФЫЎЗжзгЃЉВЛПМТЧPBCЯТЕФжиЕўаЇгІЃЌПЩФмЛсЕМжТЃК
ЦЦЛЕЯЕЭГЕФУмЖШКЭОљдШадЃКжБНгвЦГ§ПЩФмЪЙЕУЯЕЭГдкБпНчДІБфЕУЯЁЪшЛђВЛОљдШЃЌгАЯьФЃФтНсЙћЕФзМШЗадЁЃ
КіТдживЊЕФЮяРэЯжЯѓЃКPBCЯТЕФЗжзгПЩФмГіЯждкЖдВрБпНчЃЌжБНгвЦГ§ЛсКіТдетжжЯжЯѓЃЌЕМжТФЃФтНсЙћЦЋРыецЪЕЮяРэЧщПіЁЃ
removeSolventBoxOverlapЃЈЃЉКЏЪ§ЪЧШчКЮзіЕФЃП
ОпЬхЙ§ГЬМћЩЯЪіСїГЬЭМЃЌЦфжївЊзіЕФЪТЧщОЭЪЧЖдФПБъКазгЕФЭтЕФЫЎЗжзгНјааPBCДІРэЃЌНЋГЌГіФПБъКазгЗЖЮЇЕФЗжзггГЩфЛиКазгФкЃЌШЛКѓдйЭЈЙ§ОрРыРДХаЖЯжиЕўЕФШмМСЗжзгЃЌВЂНЋЦфвЦГ§ЁЃ
ЮЊЪВУДашвЊЯёremoveSolventBoxOverlapЃЈЃЉКЏЪ§етбљЃП
БЃГжЯЕЭГЕФЮяРэвЛжТадЃКЭЈЙ§НЋЫљгаЗжзггГЩфЕНФПБъКазгФкЃЌШЗБЃдкФЃФтжаПМТЧЕНPBCаЇгІЃЌЪЙЕУБпНчДІЕФЗжзгПЩвде§ШЗЕиЯрЛЅзїгУЁЃ
БмУтВЛКЯРэЕФЗжзгЗжВМЃКжБНгвЦçÍóФПБъКазгБпНчЕФЗжзгПЩФмЕМжТЯЕЭГУмЖШНЕЕЭЛђБпНчаЇгІЕФВЛзМШЗБэДяЁЃЭЈЙ§PBCДІРэКЭжиЕўМьВтЃЌПЩвдШЗБЃЯЕЭГжаЕФЗжзгЗжВМКЯРэЃЌФЃФтНсЙћИќНгНќЪЕМЪЮяРэЧщПі
3.ЭМЦЌЃКremoveSolventOutsideShellЃЈЃЉЃЌИУКЏЪ§ИљОнИјЖЈЕФАыОЖЃЈ-rshellЃЉвЦГ§ШмМСЗжзгЁЃЫќНЋШЅГ§ЫљгаОрРыШмжЪЗжзгГЌЙ§жИЖЈАыОЖЕФШмМСЗжзгЁЃ
❝
ИУЙ§ГЬЭЌremoveSolventBoxOverlapЃЈЃЉжаЕФЙ§ГЬРрЫЦЃЌВЂВЛЪЧжБНгвЦГ§ЫЎЗжзгЃЌЖјЪЧПМТЧСЫPBCЃЌетРяВЛдйзИЪіЁЃ
4.ЭМЦЌ:removeSolventOverlappingWithSoluteЃЈЃЉЃЌИУКЏЪ§гУгквЦГ§гыШмжЪЗжзгжиЕўЕФШмМСЗжзгЁЃЫќЭЈЙ§МьВщШмжЪКЭШмМСЗжзгжЎМфЕФОрРыРДЪЖБ№КЭвЦГ§етаЉжиЕўЕФЫЎЗжзгЁЃ
❝
1.ИУЙ§ГЬЭЌremoveSolventBoxOverlapЃЈЃЉжаЕФЙ§ГЬРрЫЦЃЌВЂВЛЪЧжБНгвЦГ§ЫЎЗжзгЃЌЖјЪЧПМТЧСЫPBCЃЌетРяВЛдйзИЪіЁЃ
2.removeSolventBoxOverlapЃЈЃЉКЭremoveSolventOverlappingWithSoluteЃЈЃЉХаЖЯжиЕўЕФВЮЪ§жївЊРДдДгк-radiusКЭ-scaleбЁЯюЃЌвдМАЪ§ОнЮФМўvdwradii.datжаЕФЗЖЕТЛЊАыОЖаХЯЂЃЌГЬађгХЯШЖСШЁvdwradii.datжаЕФЗЖЕТЛЊАыОЖЃЌВЂАДее-scaleЫѕЗХетаЉАыОЖЃЌЖдгкЖдЫЎжаЕФЕААзжЪЃЌЪЙгУФЌШЯжЕ0.57ПЩвдЕУЕННгНќ1000 g/lЕФУмЖШжЕЁЃШчЙћЪ§ОнЮФМўжаевВЛЕНЫљашЕФАыОЖжЕЃЌЯргІЕФдзгНЋЭЈЙ§-radiusбЁЯюРДЩшЖЈОрРыЁЃ
5.ЭМЦЌЃКremoveExtraSolventMoleculesЃЈЃЉ,ИУКЏЪ§гУгквЦГ§ЖюЭтЕФШмМСЗжзгЁЃШчЙћЪЕМЪШмМСЗжзгЪ§ГЌЙ§жИЖЈЕФзюДѓжЕЃЈmax solЃЉ,ЫќЛсЫцЛњвЦГ§ЖргрЕФШмМСЁЃ
Reference
[1]
Lennard-Jones, J. E.ЃЈ1924ЃЉ. On the determination of molecular fields. Proceedings of the Royal Society of London. Series A, Containing Papers of a Mathematical and Physical Character, 106ЃЈ738ЃЉ, 463-477.ЃКhttps://royalsocietypublishing.org/doi/10.1098/rspa.1924.0082
[2]
Bernal, J. D., & Fowler, R. H.ЃЈ1933ЃЉ. A Theory of Water and Ionic Solution, with Particular Reference to Hydrogen and Hydroxyl Ions. The Journal of Chemical Physics, 1ЃЈ8ЃЉ, 515-548.ЃКhttps://pubs.aip.org/aip/jcp/article-abstract/1/8/515/177898/A-Theory-of-Water-and-Ionic-Solution-with?redirectedFrom=fulltext
[3]
Berendsen, H. J. C., Postma, J. P. M., van Gunsteren, W. F., & Hermans, J.ЃЈ1981ЃЉ. Interaction models for water in relation to protein hydration. In Intermolecular ForcesЃЈpp. 331-342ЃЉ. Springer, Dordrecht.ЃКhttps://link.springer.com/chapter/10.1007/978-94-015-7658-1_21
[4]
Jorgensen, W. L., Chandrasekhar, J., Madura, J. D., Impey, R. W., & Klein, M. L.ЃЈ1983ЃЉ. Comparison of simple potential functions for simulating liquid water. The Journal of Chemical Physics, 79ЃЈ2ЃЉ, 926-935.ЃКhttps://pubs.aip.org/aip/jcp/article-abstract/79/2/926/776316/Comparison-of-simple-potential-functions-for?redirectedFrom=fulltext
[5]
orgensen, W. L., & Madura, J. D.ЃЈ1983ЃЉ. Temperature and size dependence for Monte Carlo simulations of TIP4P water. Molecular Physics, 56ЃЈ6ЃЉ, 1381-1392.ЃКhttps://www.tandfonline.com/doi/abs/10.1080/00268978500103111
[6]
Mahoney, M. W., & Jorgensen, W. L.ЃЈ2000ЃЉ. A five-site model for liquid water and the reproduction of the density anomaly by rigid, nonpolarizable potential functions. The Journal of Chemical Physics, 112ЃЈ20ЃЉ, 8910-8922ЃКhttps://pubs.aip.org/aip/jcp/article-abstract/112/20/8910/294328/A-five-site-model-for-liquid-water-and-the?redirectedFrom=fulltext
[7]
Caldwell, J., & Kollman, P.ЃЈ1995ЃЉ. Structure and Properties of Neat Liquids Using Nonadditive Molecular DynamicsЃКWater, Methanol, and NЉ\Methylacetamide. The Journal of Physical Chemistry, 99ЃЈ16ЃЉ, 6208-6219ЃКhttps://pubs.acs.org/doi/10.1021/j100016a067
[8]
Berendsen, H. J. C., Grigera, J. R., & Straatsma, T. P.ЃЈ1987ЃЉ. The missing term in effective pair potentials. The Journal of Physical Chemistry, 91ЃЈ24ЃЉ, 6269-6271.ЃКhttps://pubs.acs.org/doi/10.1021/j100308a038
[9]
Ponder, J. W., & Case, D. A.ЃЈ2003ЃЉ. Force fields for protein simulations. Advances in Protein Chemistry, 66, 27-85.ЃКhttps://pubmed.ncbi.nlm.nih.gov/14631816/